Alpha Sarcoglycanopathy in a Turkish Family
Limb girdle muscular dystrophy type 2D (LGMD-2D) is an autosomal recessive muscular disease caused by genetic defects in sarcolemmal alpha sarcoglycan (α-SGC) glycoprotein. A 5-year-old girl applied to the pediatric neurology clinic with the complaint of weakness in lower limbs and fatigue. Her two brothers were clinically diagnosed as Duchenne muscular dystrophy without biopsy and genetic testing. Her neurological examination was normal. Her serum creatin kinase level was 13630 IU, EMG presented myopathic changes. Muscle biopsy showed dystrophic changes such as abnormalities in myofiber shape and size, interstitial fibrosis, regeneration and degeneration. Beta and delta sarcoglycans were diffusely present but gamma- and alfa-sarcoglycans could not be demonstrated in the repetative sarcoglycan staining. Also dystrophin stained positive. A homozygous R24H (c.G101A) mutation in alpha sarcoglycan gene, which is compatible with LGMD Type 2D, was demonstrated. Her misdiagnosed brothers were carried out the same mutation. Their female cousin presented with similar findings and genetic test reveal same mutation too. In this report we impressed that; muscular biopsy and molecular analysis should be performed on a female patient with hyperckemia and muscle weakness.
Keywords: LGMD type 2D; Adhalin gene; Alpha sarcoglycan; Muscular Dystrophy
Limb girdle muscular dystrophy type 2D (LGMD-2D) is an autosomal recessive muscular disease caused by genetic defects in sarcolemmal alpha sarcoglycan (α-SGC) glycoprotein. Alpha- sarcoglycan gene is encoded on chromosome 17q21. The 50k-Da protein, originally called adhalin is expressed only in skeletal and cardiac muscles. LGMD-2D has a very heterogeneous phenotype. Phenotype varies from a severe Duchenne to mild Becker phenotype. LGMD-2D predominantly affects proximal muscles around the scapular and the pelvic girdles [1]. The age of onset, rate of progression, and the severity of disease can vary between and also within affected families. The most clinically severe course is generally observed when the sarcolemmal α-SGC is totally absent whereas milder phenotypes are observed when residual proteins are present. Symptomatic cardiomyopathy is rare. Serum creatine kinase levels are usually over 5000 IU. The differential diagnosis for LGMD-2D includes Duchenne and Becker muscular dystrophies (DMD/BMD) and it is impossible to reach a diagnosis on clinical grounds alone. Therefore, immunohistochemical staining of a muscle biopsy and molecular genetic analysis are mandatory for the correct diagnosis [2,3].
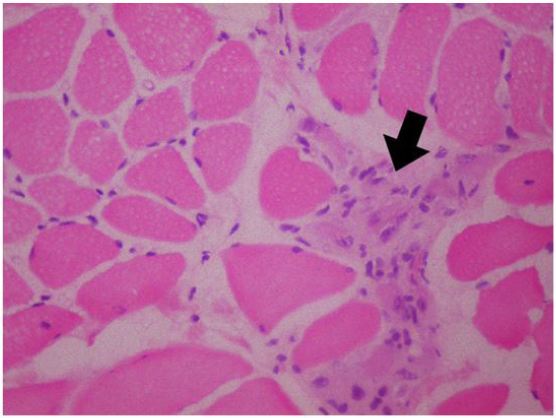
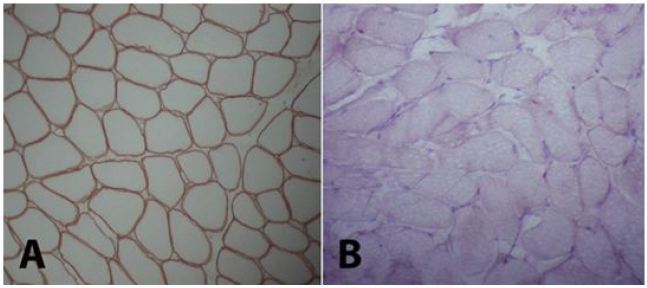
A 5-year-old girl who has no remarkable illness in her history, applied to the pediatric neurology clinic with the complaint of weakness in her lower limbs and fatigue. Her two brothers (8 and 12 years old) have diagnosis of Duchenne muscular dystrophy withoutconsulting to biopsy and genetic testing. Her neurological examination was normal, no pseudohypertrophy seen, patella reflexes were active, muscular strength was (evaluated with Medical Research Council (MRC) Scale for Muscle Strength) 5/5 in proximal and distal parts of all four limbs. Gower’s sign was negative. In biochemical tests her serum creatine kinase (CK) level was 13630 IU (normal <200IU). The needle EMG examination had shown myopathic changes that were short-lasting motor unit potentials and short amplitudes -which are the most common findings of myopathies- while sensory and motor nerve conductions were normal. Muscle biopsy was performed, because our patient has woman gender and her brothers has myopathic findings too. Histopathological evaluation was performed on a Gastrocnemius muscle biopsy specimen. The muscle biopsy specimen was frozen in isopentane precooled to -160 °C in liquid nitrogen. Cryosections were immunostained for dystrophin with a polyclonal antibody (NeoMarkersTM Leica Biosystems. USA) using a monoclonal spectrin antibody (NovocastraTM Leica Biosystems. USA) as a control. A myosin heavy chain neonatal (neonatal myosin, NovocastraTM Leica Biosystems TM Leica Biosystems. USA) antibody was used to identify pathological immature fibers. SGCs were detected with anti a-, b-, d-, and g-SGC antibodies (Novocastra, UK, NCL-a-sarc for alpha SGC and Novocastra, UK, NCL-b-d-g-sarc for beta, delta, gamma SGC). Muscle biopsy showed dystrophic changes such as abnormalities in myofiber shape and size, interstitial fibrosis, regeneration and degeneration. Beta and delta sarcoglycans were diffusely present but gamma- and alfa-sarcoglycans could not be demonstrated in the repetative sarcoglycan staining. Also dystrophin stained positive (Figure 1 and 2). This findings are compatible with Limb-Girdle Muscular Dystrophy type 2 (LGMD2). Genetic tests were performed to make a differential diagnosis between type 2D and 2C. Genomic DNA was extracted from whole peripheral blood using a commercial DNA extraction kit (Qiagen GmBh, Germany) following the standard manufacturer’s protocol. The concentration of the sample DNA was determined by a NanoDrop spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). Exon regions and the flanking short intron sequences of the SGCA gene were amplified using polymerase chain reaction. A homozygous R34H (c.G101A) mutation in alpha sarcoglycan gene, which is compatible with LGMD type 2D, was demonstrated.
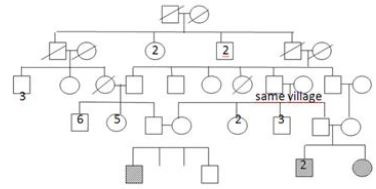
It appears that her two brothers were diagnosed Duchenne muscular dystrophy without consulting to genetic tests and biopsy in a health center. They were included in the clinical follow- up protocol. Physical examination of the older sibling (12-year-old) showed that he couldn’t walk and was obliged to use a wheelchair. Patella and Achilles reflexes were absent. Gastrocnemius pseudohypertrophy and scapular winging were significant. Muscle strengths in the proximal, and distal part of the lower (2/5, and 2-3/5) and the upper limbs (3/5, and 3/5) were measured. Serum CK level was 4500IU. Other sibling, who was 8 years old was presented with gait disturbance 86 and had difficulty to climb stairs. He had significant sacral lordosis, gastrocnemius hypertrophy. Patella and achilles reflexes were absent. Serum CK level was 25500 IU. Muscle biopsy and genetic tests were also made in two boy siblings. Results were the same with the 5-year-old girl patient, so three siblings had the diagnosis of LGMD type 2D. Parents confirmed that they are heterozygous carriers of the mutation. Ten months later their female cousin presented with similar findings. Pedigree of the family presented in the Figure 3. Genetic tests reveal the same homozygous mutation.
Limb-girdle muscular dystrophy (LGMD) is a term for a group of diseases that cause weakness and wasting of the proximal muscles in the arms and legs, specifically the muscles of the shoulders, upper arms, pelvic area, and thighs. Any male or female suspected of having limb-girdle muscular dystrophy must first be evaluated for dystrophinopathies. Duchenne muscular dystrophy (DMD) usually presents in early childhood and is rapidly progressive, with affected children being wheelchair-bound by age 12 years [4]. Few survive beyond the third decade. LGMD type 2A,2I and 2D can mimic DMD with calf pseuohypertrophy, waddling gait and difficult climbing [6].
LGMD2D is the most common sarcoglycanopathy worldwide and it is seen in the absence of adhalin protein which is encoded in chromosome 17q21. Frequently serum CK level is over 5000 IU. Significant gastrocnemius hypertrophy and scapular winging are common, like Duchenne muscular dystrophy. Therefore, genetic tests and muscle biopsy are necessary for differential diagnosis. LGMD2D has a broad spectrum with cases similar to that we present in the report. Phenotype varies from a severe Duchenne to mild Becker phenotype in late adolescents [3,5]. Patients with any of the LGMDs may be clinically indistinguishable from those with the primary dystrophinopathies. It is likely that the prevalence of LGMD is underestimated and a number of male patients are incorrectly diagnosed with DMD or BMD [6]. Just like LGMD 2D, there are reports that LGMD 2I can show Duchenne-like physical examination findings. Scwartz, et al. found homozygos mutations in 9 of 102, heterozygos mutations 4 of 102 male patients who were deletion/duplication-negative for dystrophin gene. A definitive diagnosis rests on performing the appropriate immunohistochemical examination as well as doing a molecular analysis [7]. A normal dystrophin staining pattern should be seen as well as an autosomal recessive mode of inheritance. In contrast, the patients with dystrophinopathies may show variable findings from a normal to a regional absence or a mosaic pattern of sarcolemmal staining with anti-SGCs antibodies which correspond to an abnormal organization of the cell-membrane-associated dystrophin glycoprotein complex. Therefore, it is necessary to perform a careful examination of the immunohistochemical staining as well as a genetic study in order to make the correct diagnosis. At the onset symptoms of all siblings espe 115 cially involved lower limbs when they were 5 years of age. Serum CK levels increased over 20 times of normal. The 12-year-old patient’s CK was not over 20 times of its normal range (normal <200 IU). It was probably caused by muscle wasting. First-degree consanguinity was detected.in patient’s parents which indicated that this muscular dystrophy was of inherited autosomal recessive type. Electrophysiological examinations demonstrated significant myopathic findings like short lasting motor unit potentials at short amplitude and denervation potentials such as positive waves and fibrillation potentials in all siblings. Muscle biopsy showed significant dystrophic features. The sarcolemmal expressions of dystrophin, beta and delta sarcoglycans were diffusely present, while alfa- and gamma- sarcoglycans were not stained which was compatible with LGMD Type 2 [1]. A homozygous R34H (c.G101A) mutation in alpha- sarcoglycan gene (SGCA) was found, which was compatible with LGMD type 2D. In the new era, genetic studies have discovered previously unknown mutations [7]. The lower baseline expression of SGCA in cardiac muscle probably explains the less severe cardiac dysfunction in patients with LGMD type 2D and the absence of mental retardation can be explained by the baseline absence of adhalin expression in the brain [9]. Corticosteroid treatment and gene therapy are on trial [10].
LGMD type 2D presents with typically Duchenne muscular dystrophy symptoms therefore, without muscle biopsy and genetic testing it can be confused with Duchenne muscular dystrophy as is seen in our patients [11]. We describe three siblings with limb-girdle muscular dystrophy type 2D with a homozygous R34H (c.G101A) mutation in alpha-sarcoglycan gene (SGCA). These patients 138 illustrate that the differential diagnosis of muscular dystrophies is impossible with clinical findings alone. Therefore, a muscle biopsy and DNA analysis remain essential methods for diagnosis of muscle diseases.
In conclusion, in a male patient presenting with hyperCKemia and muscular weakness first we must perform genetic analysis for Duchenne disease, then we can consider muscle biopsies and molecular analysis for other muscular dystrophies. In a female patient Duchenne muscular dystrophy is very rare because it’s an X-linked condition. Therefore muscular biopsy and molecular analysis should be performed on a female patient with hyperckemia and muscle wasting.