Anomalous Left Main Coronary Artery Origin from Right Pulmonary Artery with Preserved Left Ventricular Systolic Function and Myxomatous Degeneration of Mitral Valve
Anomalous origin of the left coronary artery from the pulmonary artery (ALCAPA) syndrome is a rare congenital anomaly. It affects 1 of every 300,000 live births. Up to 90% of patients with ALCAPA syndrome die within the 1st year of life due to “coronary steal” phenomenon. We present a case of 8-year-old girl with anomalous origin of left main coronary artery from right pulmonary artery with preserved left ventricular systolic function, associated with myxomatous degeneration of mitral valve and severe MR.
Keywords: ALCAPA Syndrome; Anomalous Coronary Arteries; Mitral Regurgitation; Mitral Valve Prolapse
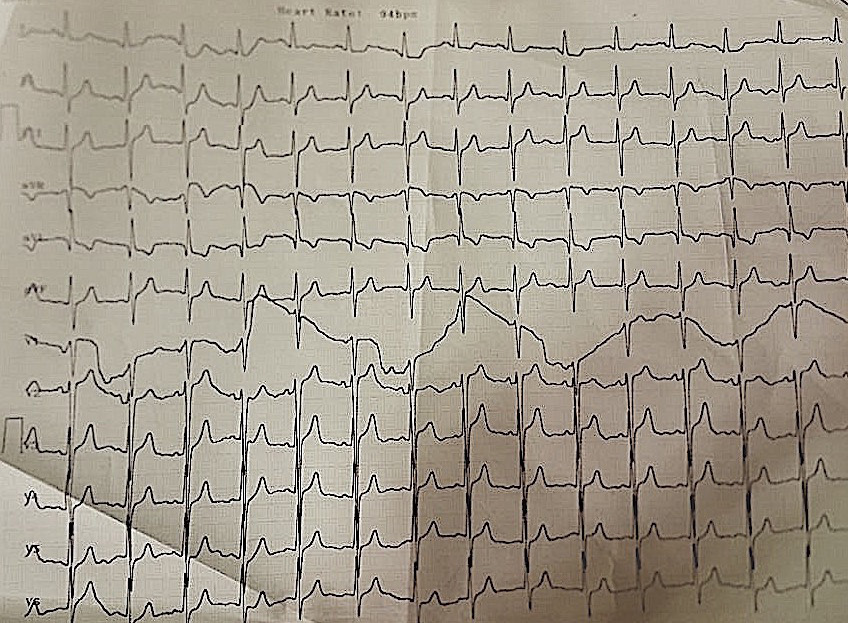
Eight-year-old girl presented with dyspnea NYHA (New York heart association functional classification) class II. On clinical examination, she had normal development with mild precordial bulge, localized apex visible and palpable at left 5th intercostal space outside the midclavicular line, with soft pan systolic murmur grade 5 radiating to axilla. Her ECG (electrocardiography) showed normal sinus rhythm with heart rate 80 bpm, normal axis and normal QRS duration with preserved R waves in anterior and inferior leads (Figure 1 and 2).
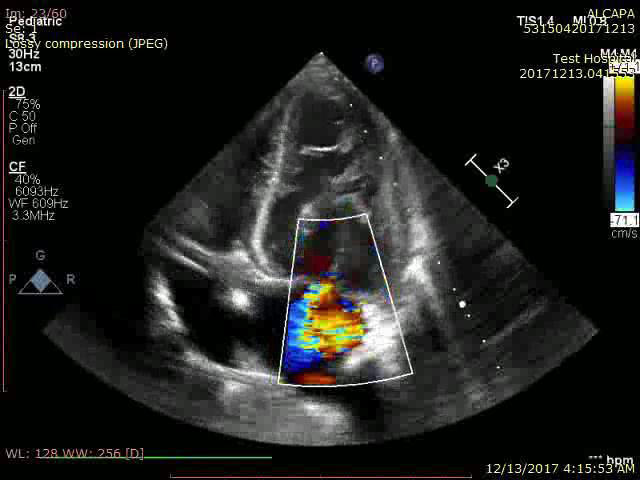
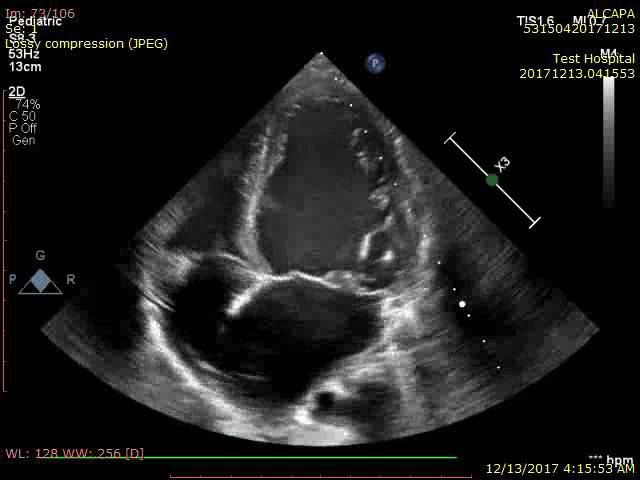
Her transthoracic echocardiography showed dilated LV (left ventricle) 45x31mm with preserved systolic function. Estimated ejection fraction 62% by m mode, 60% by modified Simpson method with no resting SWMA (segmental wall motion abnormality) visualized (Video 1). Myxomatous degeneration of MV (mitral valve) with prolapsing both leaflets and severe mitral regurgitation (Figure 3 and Figure 4) (Video 2). Continuous flow inside interventricular septum in apical 4 chamber view was noticed (video 3) with reversal of flow inside dilated left coronary artery in PSAX (parasternal short axis) view (Video 4). Normal right sided structures. Normal estimated PAP (pulmonary artery pressure). Normal aortic valve morphology and flow with no aortic coarctation.
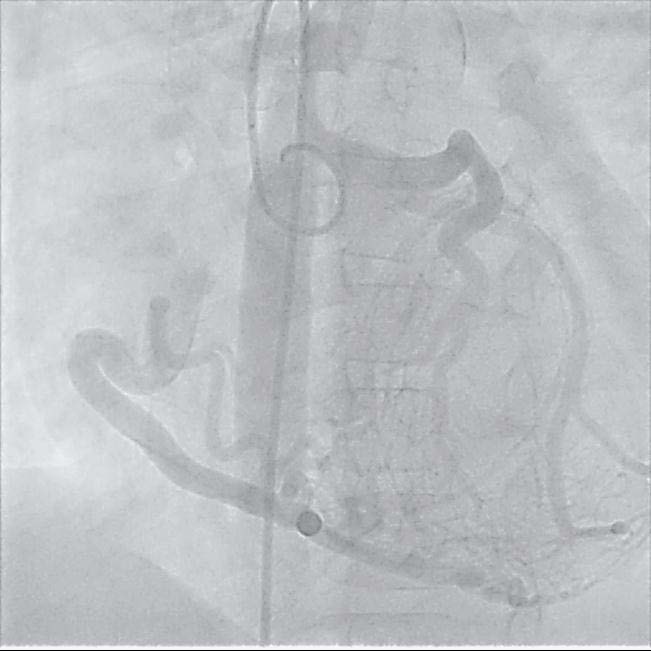
Diagnostic cardiac catheterization was done. Aortic root injection in LAO (left anterior oblique) and RAO (right anterior oblique) views, showed dilated RCA (right coronary artery) filling LAD (left anterior descending artery) through significant mesh of collaterals, followed by filling of pulmonary artery (Figure 5) (Video 5) Patient was referred to surgery and underwent coronary button transfer technique of left main coronary artery into ascending aorta, as well as mitral valve repair. Where left main coronary artery was found originating from right pulmonary artery in a course very near to aortic root.
In infant type of ALCAPA syndrome if there is little or no coronary collateral development, the onset of symptoms usually occurs about 8 weeks after birth. When the reversal of flow in the LCA (left coronary artery) occurs, a limited blood supply to the left ventricular myocardium leads to congestive heart failure and mitral insufficiency secondary to myocardial infarction. Without surgical repair, mortality may reach up to 90% of patients within weeks or months of birth Patients with ALCAPA syndrome who develop significant collateral circulation from the RCA to the LCA, survive beyond infancy [1,2]. However, collaterals may not be sufficient to supply the left ventricle, especially in the subendocardial region leading to chronic left ventricular subendocardial ischemia. Resulting in malignant ventricular dysrhythmias and sudden cardiac death in 80-90% of cases [3-7]. Patients may be asymptomatic, or they may present with mitral insufficiency, ischemic cardiomyopathy, or malignant dysrhythmias, which lead to sudden death [1]. Our patient presented at age of 8 years, with dyspnea NYHA class II mostly related to severe mitral insufficiency. She was clinically compensated, with no previous symptoms or signs suggestive of heart failure. She had no palpitations, no previous syncope, nor presyncope. Her angiography showed significant collateral circulation from RCA to LCA. Which justify her preserved left ventricular systolic function in ALCAPA syndrome, the LCA typically arises from the left inferolateral aspect of the main pulmonary artery just beyond the pulmonary valve. Then it courses toward the interventricular groove and branches into the left anterior descending (LAD) and circumflex arteries. An isolated anomalous origin of the RCA, circumflex artery, and LAD artery from the pulmonary artery also has been described. However, in more than 90% of cases, the LCA originates anomalously from the pulmonary artery, in our case left main coronary artery originated from right pulmonary artery in a coarse very near to aortic root [8].