Bifurcation of Aorta: A Diagnostic Challenge and Associated Infectious and Congenital Anomalies
Double aortic arch is a rare cardiovascular anomaly. It represents about 61% of vascular rings, found in both gender equally, and results from persistence of the embryonic fourth arch. It is usually discovered at postmortem or at autopsy in the dissecting room. Most of the recorded cases had symptoms of constriction of the esophagus and tracheas, resulting in death from broncho-pneumonia at an early age, but sometimes patients with this condition have survived till adulthood. It must be considered if an infant presents with noisy breathing, persistent respiratory symptoms of infection, emesis, choking, and dysphagia or acute apneic spells. Here, we document a rare case, a 6-month-old male patient with a history of repeated admissions and referrals since birth. The characteristic complaints included attacks of respiratory distress, lower respiratory tract infections, stridor, feeding problems, vomiting, and sepsis. With a persistent right aortic arch that constricts esophagus and trachea, repeated infections, hypocalcemia, and umbilical hernia supporting 22q11 deletions disorder is being presented for the first time in Pakistan pediatric population.
Keywords: Double Aortic Arch; Respiratory Distress; Constriction; Umbilical Hernia
Vascular rings occur in many different anatomical variations and mostly result from an abnormal regression of the embryonic aortic arch during development [1]. The presence of a left or right aortic arch depends upon whichever dorsal aorta persists. If left dorsal persists, one has a left aortic arch and vice versa. If both persist, a double aortic arch (DAA) results [2]. DAA is the most common cause of tracheoesophageal compression and has a reported incidence of 46% to 76% among all form of vascular rings [3]. It is a type of vascular ring in which the ascending aorta bifurcates and forms a right and left sided arch. The two arches then encircle the trachea and esophagus and then come together distally to complete the ring and form the descending aorta [1]. The chest x-ray may be normal [4]. In most situations, the right arch is dominant, or larger than the left. It can be isolated, or part of CHARGE/VACTERAL associations or 22q11 deletions The presence of vascular rings must be considered in any infant with stridor and episodic respiratory distress, feeding rejection and multiple admission [5,6]. Other congenital anomalies, like an umbilical hernia and hypocalcaemia, septicemia, may also have an association with severe DAA as presented in this case.
A 6-month-old baby admitted in pediatrics outpatient department of Ayub Teaching Hospital, Abbottabad with repeated attacks of apnea soon after birth. He had visited the outpatients with complaints of a cough, stridor, excessive crying, respiratory difficulty and projectile vomiting associated with feeds. The child was admitted thrice for lower respiratory tract infections and once it was associated with septicemia. The examination was normal except for a small reducible umbilical hernia. Signs of respiratory distress such as tachypnea and added sounds were present in the chest when admitted with lower respiratory tract infection. Differentials of tracheoesophageal fistula, GERD, pyloric stenosis and congenital heart defects were made and investigations proceeded. Serum calcium was low 1.57 mmol/l (1.0-2.4 mmol/l), complete blood counts revealed, TLC counts 11.6 X 109/l (6-18 X 109/l), hemoglobin levels 10.6 g/dl (11.1-14.1 g/dl), MCV 81.2 fl (70-86 fl), MCH 26.6 pg (24-30 pg), MCHC 32.8 g/dl (30-36 g/dl), platelet counts were normal, however, CRP was positive. TLC counts raised up to 15.6 X 103/μl with 84% neutrophils and ALT levels raised up to 63 IU/l. Blood culture revealed the growth of Staphylococcus epidermidis sensitive to clindamycin, linezolid, and all aminoglycosides.
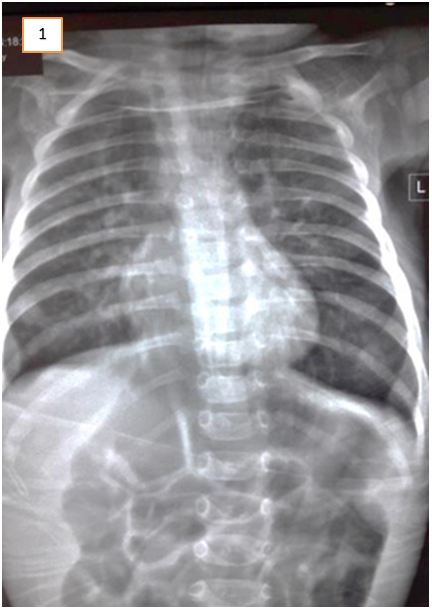
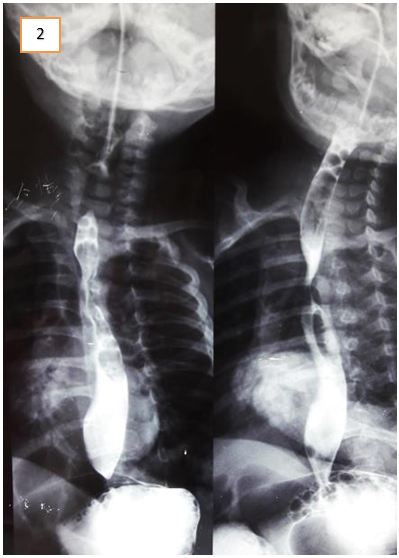
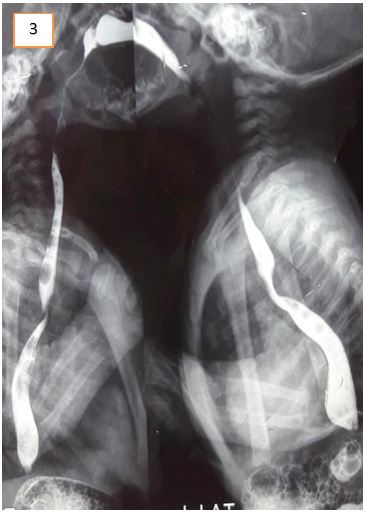
Abdominal ultrasound excluded gastric outflow obstruction. Echocardiography showed a normally functioning heart with no congenital defects. A barium swallow was done to exclude esophageal stenosis and showed narrowing of the esophagus at about T5 level. It was noted on fluoroscopy that there was a persistent mild degree of smooth narrowing at the mid-thoracic part of the esophagus with some stasis of contrast at the lower end of the esophagus. No fistulous communication was present between the trachea and esophagus (Figure 1, 2 and 3). Fluoroscopy ruled out tracheoesophageal fistula as a cause for the patient’s symptoms. After cardiothoracic consultation, external compression on the esophagus was suspected.
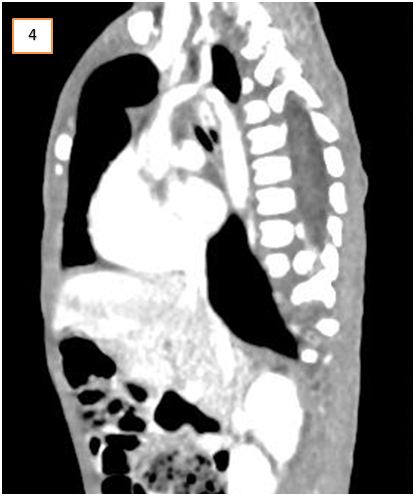
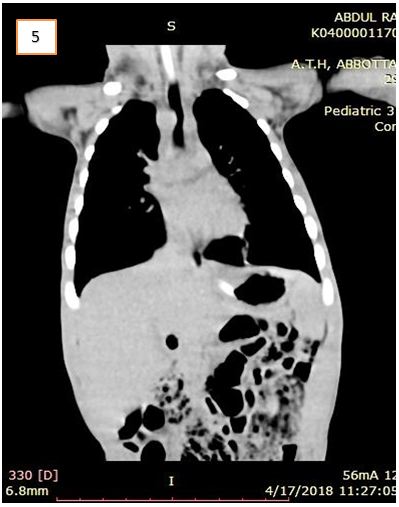
CT- chest revealed the presence of a congenital vascular anomaly of the aorta. Diagnosis of DAA was confirmed that lead to symptoms of vomiting and respiratory distress (Figure 4 and 5).
It was observed that though the ascending aorta arose normally, however, bifurcated into right and left branches instead of the aortic arch. The two aortic branches encircled the trachea and esophagus with significant compression over them just above the carina. They bifurcated into two branches bilaterally, i.e. the common carotid and the subclavian artery. Minimal bronchiectasis and fibrotic bands were seen bilaterally in lower lobes of both lungs, most probably secondary to repeated aspirations. Rest of the findings was normal. The DAA repair surgery was planned. But the child died a day before the planned operation day.
Just after birth, and based upon repeated admissions for respiratory infections, septicemia, hypocalcemia, umbilical hernia and obviously, the DAA, a final diagnosis of the presence of 22q11 deletions and Di George syndrome was made, though couldn’t be confirmed due to the demise of the child.
Double aortic arch is a rare and challenging condition for most of the pediatricians [7]. It has a typical clinical presentation but its early recognition, management, and association with CHARGE, VACTERAL, and 22q11 deletions is very rarely reported [5]. We encountered a similar case of DAA and emphasized upon the prevention of any infection, prompt diagnosis, and early management in suspected cases. As well as, its association with an umbilical hernia may be considered in some cases.
The DAA is a rare congenital anomaly of the aortic arch. In this, the two arches form a complete vascular ring that compresses both the trachea and the esophagus. Obstruction is severe in some patients. In most patients, the right arch is dominant and posterior to the trachea and esophagus while the left is hypoplastic and in front of them. The left arch is dominant in approximately 20% of cases. The two arches then join and form the descending aorta (more commonly) on the left side, right side or in the midline. The recurrent laryngeal nerves of either side usually hook around their respective aortic arches [8]. Sometimes the vascular ring is incomplete [9].
Respective common carotid and subclavian arteries arise from each aortic arch. Occasionally, the right or more commonly the left arch is atretic which is often not visible with current imaging techniques [10]. Patients present mostly signs and symptoms of the respiratory system. Most common is wheezing, followed by stridor, pneumonia, recurrent upper respiratory tract infections, respiratory distress, cough, and cyanosis [5].
DAA should be considered in any newborn or young with recurrent episodes of stridor, respiratory distress or gastrointestinal symptoms after birth. The only treatment is relief by surgical repair.
DAA is either isolated or part of CHARGE (posterior coloboma, heart defect, choanal atresia, retardation, genital, and ear), VACTERAL (vertebral, anal, cardiac, tracheal, esophageal, renal, and limb) anomalies [11]. It has also been reported in association with other chromosomal anomalies, such as 22q11 deletion, trisomy 21or other syndromes [12]. Patients should also be tested for 22q11 deletion which is associated with DAA.
After surgery, there is some relief of tracheal compression but complete normalization may take time [13]. Especially, longstanding compression that start during fetal life affects the normal development of the tracheal cartilage in the compressed segment. This leads to tracheal cartilage malacia and some stenosis. Therefore, for complete recovery patient needs to be stabilized with a temporary tracheotomy [14]. The airway obstruction may be worse in the first postoperative weeks. Most patients need continuous positive airway pressure by appropriate flows of humidified oxygen and air and are extubated soon after surgery but some require intubation and mechanical ventilation for several days [15]. A few patients have immediate relief of stridor while in others dysphagia may persist for some time.
In a review study, 81 patients of DAA, aged < 18 years, concluded with excellent postoperative outcomes, however, some respiratory symptoms persisted. Mean age of presentation in the study was 5 months and it was mentioned that most patients presented with respiratory symptoms and stridor while gastrointestinal symptoms were less common [14]. This is somehow in accordance with our case. A similar case reported recurrent respiratory tract infection, multiple admission and hiatal hernia with the gastroesophageal reflux disease. This patient chest x-ray was also normal [15]. Other supportive data is lacking here.
The common risks of open surgical repair of DAA are an injury to the recurrent laryngeal nerve, causing vocal cord paralysis. Injury to the lymphatic system leading to postoperative chylothorax. Additional risks include injury to the lung, hemorrhage requiring blood transfusions and wound infection [16,17].
Primarily, X-ray chest is unable to detect aortic anomalies. Secondly, although rare and considered a diagnostic challenge, but DAA is an important cause of repeated respiratory tract infections and feeding problems soon after birth. It should be considered as a possible cause in these children, as early recognition and management can prevent death. Due to Association of this anomaly with CHARGE, VACTERAL, and 22q11 deletions, these should be considered in the diagnosis as well. Proper vaccination, good hygiene, safe feeding and prompt visits to a physician is highly recommended for the prevention of recurrent infection/sepsis and early surgical repair to relieve the symptoms.