Cancer Stem Cells in Tumors Arisen from Digestive Organs
The digestive system encompasses several organs in which tumors occur with an elevated prevalence and mortality worldwide. Increasing evidence shows that cancer stem cells (CSCs) may play a critical role in tumor development and progression. In addition, CSCs are characterized by resistance to conventional therapies probably explaining why is difficult to completely eradicate cancer. Thus, the research on CSCs is critical to develop therapeutic strategies for a more effective reduction in the risk of relapse and metastasis and possible total elimination of tumors. This review compiles information about markers, signaling pathways and putative targeting of CSCs in several organs of the digestive system in order to unravel common and divergent patterns and behaviors that could be useful for future CSC research.
Keywords: Cancer stem cells; Colorectal tumor; Pancreatic adenocarcinoma; Hepatocarcinoma; Gastric cancer; Esophageal tumors; CD133; LGR5; CD90
Multicellular organisms develop from pluripotent embryonic stem cells capable to differentiate into all cellular types of the organism. In the adult, stem cells derived from the primitive stem cell pool for each germ layer are present in all tissues and operate as a cellular reservoir that fuels homeostatic cell turn over and readily promotes tissue regeneration after injury or stress. This description implies that tissue stem cells exhibit self-renewal and multilineage differentiation potential achieved by symmetric and asymmetric divisions respectively. These asymmetric divisions lead to progenitor or transient amplifying cells with more restricted lineage potential, progenitor cells eventually produce the most differentiated progenies.
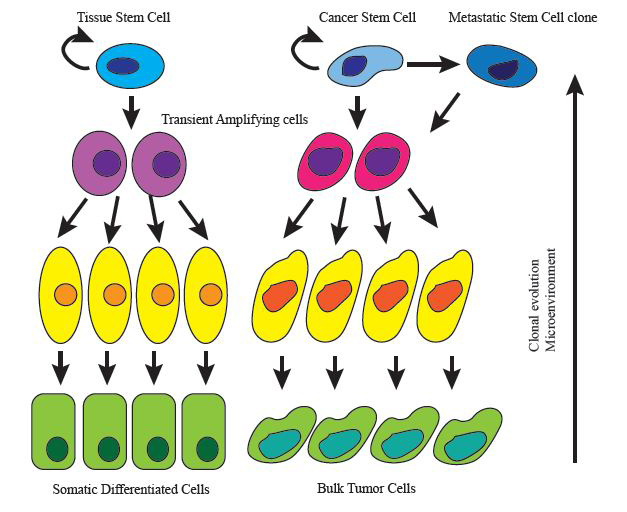
Similar to normal tissues, tumor tissues are mostly composed of heterogeneous populations of cancer cells, containing multiple cellular subtypes. One explanation for the observed tumor heterogeneity is provided by the “cancer stem cell” (CSC) model. This model intends the possibility that malignant tissues, like their normal counterparts might be organized according to a hierarchical structure akin to a stem cell system [1] as they often exhibit a physiology and cell composition vaguely resembling the tissue from which they originate ( Figure 1). According to this, tumor cells evidencing self-renewal capacity, tumor seeding potential and ability to recapitulate tumor cellular heterogeneity are designated CSC. This definition of cancer stem cells is based on functional assays that account for self-renewal in vitro and tumor initiation in vivo mostly in immunodeficient mice. In fact the cellular and molecular mechanisms that confer the special characteristics of CSCs are yet poorly understood although key embryonic and developmental pathways active in non cancerous stem cells are also described as reactivated in CSCs such as Hedgehog or Wnt pathways [2]. It is important to clarify that cancer stem cells in a tumor are not necessarily related to the cell of origin of the same tumor, which defines the normal cell within the tissue that acquires the first cancer promoting mutation [3]. Therefore, despite key properties shared between tissue stem cells and their CSCs counterparts, in the majority of tumors it is unclear if CSCs actually originate from stem cells or progenitors or differentiated cells within the organ or tissue.
The CSC model is supported by an increasing body of experimental evidence. After the pioneering identification of leukemia initiating cells in 1997 [4] followed by the characterization of breast cancer stem cells [5], malignant cells with stem cell properties have now been prospectively identified across many tumor types, both in primary human cancers and in experimental mouse cancer models. Moreover, lineage-tracing experiments have provided formal demonstration of multi-lineage differentiation as a source of epigenetic diversity in cancer tissues [6].
These epigenetic alterations as well as the microenvironment might promote the evolution of CSCs into different clones simultaneously present in the tumor, but bearing distinct invasive potential, hypoxia endurance or susceptibility to chemotherapy.
On the other hand, the CSC model has important implications for clinical oncology, as it provides a new conceptual framework for the development of prognostic biomarkers and the design of anti-tumor drugs.
The most common type of esophageal cancer worldwide is the esophageal squamous cell carcinoma (ESCC) which is the sixth leading cause of cancer related deaths [7]. Its aggressive nature, together with late diagnosis and poor knowledge of the cellular and molecular mechanisms of the disease help to explain its 5-year survival of 20-30% after curative surgery. In western countries, the predominant esophageal cancer subtype is esophageal adenocarcinoma, that is tipically preceded by Barrett´s esophagus disease, a metaplastic transformation of the esophagus squamous epithelium into gut-like columnar epithelium in response to gastroesophageal reflux.
Stem cell/progenitors in the mouse foregut are located in the basal layer of the epithelium [8,9]. In the adult, those progenitors are responsible for tissue renewal in homeostatic conditions [10]. However, the same cells are able to switch to a more proliferative state in repair conditions after wounding. The Lef/Tcf-family transcription factor Tcf3 labels those progenitors as it occurs in other stratified epithelia [11]. NGF receptor p75NTR and Sox2 are also expressed in esophageal progenitors. The levels of Sox2 are crucial for the balance between self-renewal and differentiation [12] as overexpression of this gene in the basal layer promotes hyperproliferation and cooperates with inflammation signaling (IL-6/Stat3) for the development of ESCC. These findings suggest that basal stem/progenitor cells are the cells of origin of ESCC.
There are few works supporting the existence of cancer stem cells in ESCC. First descriptions of tumor propagating cells were done in cell lines and identified p75NTR as marker for a subpopulation with enhanced drug resistance and expression of stem cell-related genes [13]. In another set of ESCC cell lines, high expression of CD44 correlated with enhanced tumorigenicity and drug resistance [14]. Using RNAseq analysis between tumor and non tumor tissue from patients, Thy-1/CD90 emerged as specific for tumor samples and labeled a subpopulation of cells from tumors and cell lines with stem cell features such as enhanced sphere formation capacity, high tumor initiation potential, increased chemoresistance and strong lung metastatic capacity [15]. In the analyzed samples, few overlapping between the expression of Thy-1 and CD44 or p75NTR was found. Interestingly, Thy-1+ cells showed upregulation of Ets-1 transcription factor and its downstream targets MMPs together with an EMT phenotype.
Based on the use of side population (SP) sorting for enriching in CSC populations, the ESCC cell line EC9706 contains a side population endowing CSC characteristics. The stem cell-related gene Bmi1 plays a role in the maintenance of those CSC properties [16]. Overexpression of the miR-203 targets this gene and impairs tumorigenicity and sphere formation of this SP population.
In the case of esophageal adenocarcinoma there is evidence for sphere forming cells in human disease cell lines identifying ALDH1 as a CSC marker [17]. ALDH1+ are more chemoresistant and form more spheres than ALDH1- cells. Subsequently, 5-FU-based chemotherapy of esophageal adenocarcinoma cells is enhanced by combination with metformin which reduces ALDH1+ cell sphere formation and tumor growth by targeting the PI3K/mTOR pathway [18].
Gastric cancer is a widespread tumor disease with a high incidence and mortality. Gastric tumors are essentially divided into histological groups: the intestinal and diffuse types. The former one is characterized by well-defined glandular structures derived from initial transformation of gastric mucosa cells and develop through sequential stages starting by chronic gastritis, atrophy, intestinal metaplasia and dysplasia [19]. Food-associated factors like excessive salt intake or low intake of vegetables and fruit are risk factors for this group of tumors. However the most clear environmental factor linked to gastric carcinogenesis is Helicobacter Pylori infection being the inflammation derived after infection the starting point for the transformation process leading to adenocarcinoma. The diffuse type of gastric cancer is characterized by single cells or small cell clumps proliferating with an undifferentiated phenotype.
The tissue cells that give rise to gastric CSC are not specified, it has been proposed that tissue stem cells within the stomach could be good candidates. In this regard, highly proliferative cells in the isthmus of gastric invaginations repopulate the mucosa that displays a turn over between 2-7 days. More recently the discovery of Troy+ cells on the bottom of the gastric invaginations expressing differentiation markers of zymogenic chief cells have been proposed as quiescent tissue stem cells with the capacity to give rise to all lineages in the gastric mucosa [20]. Interestingly, these Troy+ cells express, together with chief differentiation markers, several Wnt target genes suggesting that Wnt pathway is active in this subset, and, in analogy to what happens in colon and liver, mutations and deregulations of the pathway could lead to the development of gastric adenocarcinomas.
Another source of gastric CSCs might be bone marrow-derived cells (BMDC) [21]. These cells are characterized by multilineage differentiation potential and the capacity to migrate to tissues and organs during emergency states such as inflammation or injury. It has been demonstrated that BMDC home to and repopulate the gastric mucosa after Helicobacter felis chronic infection contributing to subsequent metaplasia and gastric carcinogenesis [22]. In a similar manner, BMDC have been suggested to contribute to the development of tumors in the setting of chronic H. pylori inflammation [23].
The classic combination of cell surface markers CD44/CD24 initially proposed for discriminating breast cancer CSCs also enriched for CSCs in patient gastric tumors and a human gastric cancer derived cell line [24]. 200 CD44+/ CD24+ cells still preserved tumor-initiating capacity in xenograft assays in comparison to 10000 CD44-/CD24- cells. In addition, CD44+/CD24+ cells showed enhanced expression of some members of the Hedgehog pathway.
In a similar way, CD44+/CD54+ cells representing the 0.1-19.7% of the cellular content in analyzed patient gastric adenocarcinomas display a 1000-fold increase in tumor initiating capacity comparing with unsorted cells although intermediate phenotypes (CD44+/CD54- CD44-/CD54+) also enriched for tumorigenic potential [25]. Circulating CD44+/CD54+ tumor cells from these patients also displayed sphere forming capacity.
Another potential marker for gastric CSC is Thy-1. This embryonic stem cell marker is enriched in sphere-forming tumor cells from human gastric tumors and Thy-1+ cells display higher in vivo tumorigenicity and recapitulate tumor heterogeneity better than Thy1- cells in single cell implantation assays [26].
Colorectal cancer is one of the most prevalent cancer types worldwide. Although it has an elevated cure rate of around 90%, it is still the leading third cause of cancer death in USA [7]. Bowel inflammation as it occurs in Crohn’disease and ulcerative colitis is a predisposing factor for developing the malignancy.
This cancer originates in the epithelial cells of the intestinal crypts of the colon or rectum. Activating mutations in the Wnt signaling pathway are the most probable starting event in the tumor process, in particular mutations in the tumor suppressor gene APC which controls β-catenin concentration. Alterations in P53, TGF-β pathway and activation of oncogenes such as KRAS, BRAF and PI3K are also commonly found in this type of cancer.
In the intestine it has been clearly demonstrated that the cell of origin of adenomas induced by aberrant activation of the Wnt pathway is the intestinal stem cell. Despite the fact that existence of such stem cells in the intestine was proposed decades ago, those cells were not clearly identified until the use of lineage tracing experiments showing that LGR5 specifically labeled intestinal stem cells able to regenerate the entire crypt units [27] and they matched with the previously named crypt based columnar cells. LGR5 is a Wnt target gene, it acts as a receptor for R-Spondins which are effective enhancers of Wnt signaling [28]. Subsequently, CD133 was shown to be expressed in those intestinal stem cells [29].
The same two groups responsible for the identification of colonic stem cells showed that loss of APC after specific Cre activation driven by LGR5 or CD133/Prominin-1 promoters initiates adenoma formation [29,30]. Importantly, loss of APC in progenitors or more differentiated cells does not lead to tumor formation. Although the cell of origin of colorectal cancer has been convincingly identified, there is some controversy about the identity of colorectal CSCs and a variety of markers have been used to identify them.
In addition to its role as marker and important player in the biology of normal colon stem cells and adenoma initiating cells, LGR5 has been proposed as a CSC marker in mice and human primary tumor cells [31-33]. LGR5+ cells express the highest levels of Dnmt3a, one de novo DNA methylase whose deletion significantly reduces tumor formation in the APC(Min/+) mouse model [34]. One regulator of LGR5 is GATA6. This transcription factor induces its expression and decreases the differentiation-promoting BMP signaling in adenoma stem cells [35]. Another crucial transcription factor in LGR5+ CSCs might be KLF5, an otherwise critical factor in the maintenance of the integrity of embryonic and induced pluripotent stem cells. Selective deletion of KLF5 in LGR5+ stem cells induces apoptosis and completely blocks adenoma formation [36].
CD133+ cells from several human tumor and metastatic samples readily developed tumors when transplanted into the renal capsule of immunocompromised mice [37]. Those tumors recapitulated the heterogeneity and morphology of the parental tumors. In addition, another contemporary study demonstrated that tumor-spheres were generated by CD133+ cells from colon cancer specimens and could be maintained through serial passages [38]. The CD133+ population would approximately account for the 2.5% of the tumor cells. Resistance to chemotherapy is a characteristic of these CD133+ CSCs [39]. The protection against apoptosis induced by drugs such as 5-FU and oxaliplatin would involve the production of interleukin-4 as an autocrine growth factor. However, there is some controversy about this marker as different epitopes used by available antibodies against CD133 target more or less restricted populations in the tumor samples and that could lead to misinterpretations of the results [40].
The triple combination of CD44/EpCAM/CD166 allowed for the identification of tumor cells with an elevated frequency of tumor xenograft generation in NOD/SCID mice [41]. Another usual marker of CSC, ALDH1 allowed for the isolation of a tumor population with tumorigenic capacity being as few as 25 ALDH1+ cells able to promote tumor growth [42].
Together with these markers several other proteins have been linked to colorectal CSCs [43]. Those include cell surface proteins such as CD24, β1-integrin/CD29 and CD26. The embryonic stem cell factors Nanog, Oct4, Sox2, Lin28, and c-Myc are associated with colorectal CSCs correlating with poor prognosis, resistance to therapy and relapse. Musashi-1, a RNA-binding protein involved in asymmetric division and expressed in the bottom of the crypts is upregulated in CD133+ tumor cells and derived spheroids [39].
Hepatocellular carcinoma (HCC) is a very frequent type of tumor and the third leading cause of cancer-related death worldwide [7]. Chronic viral infection with hepatitis B and C viruses is a main cause for developing this malignancy. Liver cirrhosis originated by chronic hepatitis C, alcoholism, aflatoxin or by other causes is a common and previous stage to HCC. The signaling pathways that have been implicated in the initiation and progression of HCC include RAF/MKK1/MAPK3 pathway, PI3K/AKT/mTOR pathway, Wnt pathway, IGF signaling, HGF/c-MET pathway, TGF-β pathway and angiogenic-related pathways [44,45]. The cellular origin of this disease is unclear as it includes heterogeneous pathologies and a diversity of genetic profiles pointing that probably HCC can originate from different cell lineages [46].
It has been described a set of single markers for defining populations enriched in HCC CSCs. These include EpCAM, CD44, CD24, CD133, OV6, ABCG2, Thy-1, CD13, DLK1 and calcium channel α2δ1 isoform5 [47-49]. Most of those markers are also associated to hepatic progenitors in healthy liver. Positive selection of HCC cell lines based on the use of those individual markers defines populations with enhanced tumorigenic potential when injected in immunodeficient mice. In primary samples of HCC only EpCAM, CD133, Thy-1 and calcium channel α2δ1 isoform5 have been confirmed as markers for CSC-enriched populations.
In tumors and cell lines expressing α-fetoprotein (AFP), EpCAM+ displays enhanced sphere formation, invasiveness and tumorigenic potential although those properties are not excluded from the EpCAM- population [50]. Activation of Wnt signaling promotes EpCAM expression. The embryonic transcription factor SALL4 is associated with poor prognosis in HCC, it is in fact expressed in AFP+EpCAM+ cells promoting their cancer stem cell features [48].
CD90/Thy1 marker is not present in AFP+ cells however it labels a population with high metastatic potential [51]. Both EpCAM+ and CD90+ cells can coexist in HCC patient samples but do not colocalized. EpCAM labels a population with higher tumor initiating capacity and hepatic epithelial stem cell features and AFP-CD90+ display a more mesenchymal vascular endothelial phenotype with abundant expression of c-kit and in consequence showed chemsensitivity to the c-kit inhibitor imatinib mesylate [47,51].
The calcium channel α2δ1 isoform5 is also a promising marker of HCC CSCs. An specific antibody recognizing this isoform (1B50-1) identifies a subpopulation of HCC cells with stem cell properties such as self-renewal, expression of stem cell-associated genes (OCT4, SOX2, NANOG and BMI1), increased invasiveness and a tumor initiating capacity 4-fold higher than channel α2δ1 isoform5 non expressing cells. Interestingly, the use of this marker in combination with EpCAM, CD13 or CD133 further increased the frequency of cancer stem cells [52].
There have been proposed several approaches for targeting HCC CSCs for therapeutic uses. The treatment with the antibody 1B50-1 that recognizes calcium channel α2δ1 isoform5 reduces CSCs activity by induction of apoptosis [52]. This is accompanied by a decrease in tumor size and lost of tumorigenicity in serial transplantations into nude mice. These effects can be further enhanced by combining this treatment with administration of doxorubicin that targets simultaneously both CSC and non CSC tumor populations.
Another therapeutic strategy focused on HCC CSCs implies the co-administration of branched chain aminoacids (BCAA) together with 5-Fluorouracil (5-FU). BCAA are required for ammonium metabolism in muscles when liver cannot accomplish that function and are already approved as a treatment for liver cirrhosis in Japan to treat hypoalbuminemia [53]. In HCC cell lines, BCAAs are able to decrease the percentage of EpCAM+ cells which also showed enhanced expression of the differentiation marker CYP3A4 and mTORC1 activity [54], suggesting that the reduction of the CSC population via mTOR pathway could sensitize a broader spectrum of the tumor cellularity to 5-FU therapy. In fact, xenografts of HCC cell lines respond better to the combined treatment than to 5-FU alone. Promoting the sensitivity of CSCs to γ-irradiation in Huh7 hepatoma cell line was accomplished by knockdown of the scaffold protein 14-3-3ζ [55]. Depletion of this protein upregulated the levels of pro-apoptotic proteins in CD133+ CSCs and significantly abolished observed apoptosis resistance in these CSCs when exposed to radiotherapy.
Pancreatic ductal adenocarcinoma (PDAC) is one of the deadliest solid cancers despite the fact that it is not a very frequent malignancy. Unfortunately the overall 5-year survival rate among patients has remained stable at around 5% over the past decades. The causes for tumor development remain unknown although smoking, diabetes and chronic pancreatitis are considered relevant risk factors [56].
The current disease model establishes an initial preinvasive state termed pancreatic intraepithelial neoplasia (PanIN) with three stages of increasing cellular atypia and mutations. PDAC is characterized by near-universal presence of mutations in KRAS2 gene (90% of all tumors) and frequent deregulation of crucial embryonic signaling pathways like Hedgehog and Notch1. Kras is considered a master regulator of pancreatic ductal adenocarcinoma initiation and progression [57].
There are well-established mouse models demonstrating a role of acinar cells, including centroacinar cells, as putative tumor-initiating cells of PDAC. Acinar-ductal metaplasia is a phenotype associated with pre-malignant adenocarcinoma that has been proposed as the earliest pancreatic lesion and, as such, precursor of low-grade PanINs. Studies using specific promoters for these cells such as Elastase or proCPA1 show that oncogenic Kras and collaborative mutations in Trp53 or Ink4A/Arf can induce adenocarcinomas when animals are simultaneously treated with caerulein, an inducer of chronic pancreatitis [58,59].
In 2007 it was presented the first evidence for CSCs in human pancreatic tumors. A PDAC subpopulation defined by a CD44+CD24+ESA+ phenotype injected into immunodeficient mice gave rise to tumors while their triple negative counterparts did not show tumorigenic capacity. In particular, as few as 102 CD44+CD24+EpCAM+ cells were sufficient to initiate tumors in 50% of transplanted mice [60]. Triple positive cells fulfill other cancer stem cell requirements such as self-renewal and generation of tumors with the same heterogeneity as the parental one. In a different study, CD133+ cells isolated from fresh human tumor samples were highly tumorigenic [61]. Tumor formation in immunodeficient mice was detectable after injecting 5x102 CD133+ cells, but no tumor growth was detected after inoculation of 106 CD133- cells. In addition, CD133+ cells were able to form spheres in serum-free non-adherent conditions and generate tumors in serial transplantation. Remarkably, only CD133+ cells expressing the chemokine receptor CXCR4 exhibit metastatic potential [61]. This finding suggests that there should be different CSCs clones within the tumors exhibiting heterogeneous capacities.
Apart from these more commonly used markers, other approaches have been proposed for the enrichment of pancreatic CSCs. In a study including 269 tumor samples, ALDH1 expression was significantly associated with poor prognosis and ALDH1+ cells were 5 to 11 times more clonogenic as compared to their negative counterparts and presented an enhanced migratory and invasive potential [62]. In human PDAC cell lines, cells with low activity of 26S proteasome displayed cancer stem cell properties [63].
In addition, the receptor for hepatocyte growth factor c-Met has also being proposed as a CSC marker in pancreatic cancer [64] and when combined with CD133 or CD44 strongly selected for cancer stem cells as demonstrated by enhanced in vivo tumorigenicity as compared to each of the single markers. On the other hand, research performed in a mouse model of the disease defines an epithelial EpCAM+CD24+CD44+CD133-Sca1- population bearing CSC properties and metastatic potential [65].
Several strategies have been proposed in order to fight against PDAC CSCs [66]. Decreasing the CSC resistance to standard chemotherapy with gemcitabine was demonstrated by the combination of gemcitabine with the c-met inhibitor XL184 [64]. This inhibitor caused a reduction in the c-Met+ pool together with decreased tumor sphere formation. It also prevented tumor growth for a longer period than gemcitabine alone.
The specific reactivation of the Activin/Nodal pathway in pancreatic CSCs has been explored as a target for eliminating CSCs and thus knock-down of this pathway reduces tumor sphere formation and virtually abrogates in vivo tumorigenicity [67]. Combination of the Nodal/Activin inhibitor SB431542 with gemcitabine and Hedgehog pathway inhibitor for targeting the stromal cells proved to be a successful therapy for long-term prevention of relapses in human tumor xenografts.
Metformin, a common drug for type II diabetes therapy displays anti-tumor activity in some malignancies. In pancreatic cancer, treatment with metformin diminished tumor sphere formation [68] and appears to act selectively in CSCs where it causes a rise in reactive oxygen species production and a fall in their mitochondrial transmembrane potential causing a deadly energy crisis [69].
As mentioned previously, key embryonic and developmental pathways are frequently essential for stem cell and CSC function. Wnt/β-catenin pathway is active in intestinal stem cells [2] and in hepatic oval cells which have been proposed as facultative tissue stem cells in the adult liver [70]. Aberrant activation of Wnt pathway in intestinal stem cells promotes the formation of adenomas [27]. Similarly, activation of the pathway in a specific subpopulation of CD44+ cells of the stomach epithelium with stem-like properties is able to induce expansion of those cells when combined with prostaglandin E2 activation eventually leading to tumor formation [71]. On the other side, blockage of the pathway affects CSC function. Expression of the Wnt inhibitor DKK1 impairs self-renewal in gastric tumorspheres [72]. In HCC cell lines, inhibition of Wnt pathway reduces the fraction of putative OV6+ CSCs and its resistance to chemotherapy [70], suggesting that the pathway is important for the maintenance of some CSC features. In some cases, this pathway is closely related to the CSC phenotype as it occurs in HCC where the CSC marker EpCAM is also regulated by Wnt signaling at the transcriptional level [73] and EpCAM+ HCC cells display enhanced Wnt pathway activity [50].
It is unclear if hyperactivation of this pathway is an intrinsic characteristic of CSCs. At least that is not the case in colon cancer where CSCs are characterized by high Wnt signaling activity. In analogy to healthy tissue, the tumor microenvironment extrinsically modulates Wnt pathway in CSCs, in particular, hepatocyte growth factor secreted by myofibroblasts surrounding CSCs activates beta-catenin-dependent transcription and subsequently CSC clonogenicity [74].
Sonic Hedgehog pathway is a classical developmental pathway frequently altered in cancer. In fact, there is increasing evidence for its relevance in malignancies developed in digestive organs. Enhanced expression of Sonic Hedgehog (SHH) has been described in gastric metaplasia and tumors. Activation of the pathway promotes motility and invasiveness in gastric tumor cells in a TGF-β receptor ALK5 dependent manner [75]. Upregulation of SHH and downstream members of the pathway such as PTCH1 and GLI has been described in gastric CD44+CD24+ CSCs in comparison with non tumorigenic CD44-CD24- cells [24]. Another study describes that inhibition of the pathway using ciclopamin reduces self-renewal in vitro and potentiates sensitivity to chemotherapy in gastric tumor spheres in comparison with adherent tumor cells [76]. Aberrant activation of SHH pathway has also been described in human pancreatic cancer lines where activated Gli genes repress Fas expression and promote the expression of survival-related genes such as Bcl-2 and PDGFRα [77].
Independently of its role in the maintenance of the epithelial integrity of tissues and its promotion of invasiveness in many tumor types, TGF-β signaling is relevant for CSC biology. For example, in agreement with defective activation of TGF-β pathway in esophageal adenocarcinoma, loss of TGF-β adaptor β2 Spectrin enhances sphere formation capacity in several human cell lines and tumor growth in immunocompromised mice [78]. Lack of β2 Spectrin promotes Notch signaling leading to upregulation of the stem cell marker SOX9 associated to acquisition of CSC properties in esophageal cancer cell lines [79]. In the liver, this pathway could also be relevant in liver adult stem/progenitor cells in order to protect them from alteration of their stemness features. Inactivation of the pathway occurs in parallel to malignant transformation of putative liver stem cells caused by IL6/STAT3 signaling activity [80]. In PDAC, activin/Nodal signaling instead of TGF-β are functionally relevant for pancreatic CSCs [67]. This embryonic pathway is not active in the adult pancreas but proved to be reactivated in CSCs where Alk4/7, the TGF-β superfamily receptors for Activin/Nodal drive self-renewal of CSCs. Importantly, there is a manifested relationship between the pathways governing CSC stemness and those implicated in epithelial-mesenchymal transition (EMT). For example, Hedgehog, Notch, and Wnt signaling pathways are key pathways for the biology of pancreatic CSCs, but also play a role in the regulation of EMT [81].
The CSC hypothesis has given new hope for better treatments in cancer malignancies. Tumors arisen in the digestive system are heterogeneous. However, inflammatory processes and metaplasias are usually frequent in the initial steps of most of them and might indicate shared cellular mechanisms beyond histological heterogeneity. The isolation of tumor populations enriched in CSCs has been reported in all these tumors and several cell surface proteins including CD133, EpCAM and Thy-1 emerge as common CSC markers independently of the tumor type. These facts suggest common phenotypes for CSCs in digestive tumors (Table 1). However it is unclear if those proteins have a functional relevance in CSC biology. Developmental pathways such as Wnt and SHH pathways together with TGF-β signaling are frequently reactivated in CSCs and participate in the control of essential mechanisms such as self-renewal and survival.
Tumors arisen from the digestive system manifest high incidence and mortality rates. Despite the advances in identifying and characterizing cancer stem cells in these tumors, CSC research in digestive-related malignancies will doubtless benefit from the identification of more specific markers that provide more pure CSC populations, as it is the case for colorectal cancer and LGR5 marker. The establishment of in vitro systems would facilitate long-term and more detailed studies on CSC biology and allow high throughput drug assays. One example of these systems is already developed in colon [82]. In addition, the use of genetic tools are still required for in vivo study of CSC behavior and interaction with the microenvironment.
Apart from adding more evidences in support of the CSC hypothesis, the identification and characterization of CSCs in these highly prevalent cancers offers new therapeutic strategies for fighting against these diseases. Nevertheless, the cross-talk between CSCs, non-CSCs and stromal components and the emerging proofs for plasticity among non-CSC and CSC states disclose that effective therapies most probably should also take into account not only CSCs but also bulk cells and the surrounding stroma.