Crouzon Syndrome - A Case Report of Rare Genetic Disorder with Review of Literature
In 1912, a French neurologist, Octave Crouzon (1874-1938) first described the crouzon syndrome and in 1937 Atkinson reviewed eighty six cases that were published and found about 67% of the cases were familial and 33% were sporadic, representing new mutations. Crouzon syndrome has a prevalence of 15-16% in one million new born and 4.5% of all craniosynostosis. Here we present a case of crouzon syndrome in 6 year old boy with review of literature.
Keywords: Crouzon Syndrome; Rare Case; Premature Synostosis; MutationCASE
Crouzon’s syndrome was first described by a French neurologist, Octave Crouzon in the year 1912 as one of the varieties of craniosynostosis. It is otherwise called as craniofacial dysostosis [1]. It has a prevelace of 15-16% in one million new born and 4.5% of all craniosynostosis. The genetic defect from mutation in the fibroblast growth factor receptor 2 (FGFR) on chromosome locus 10q25q26 which results in the early fusion of the skull bones during the development of the foetus. Cranial malformation in Crouzon syndrome depends on the order and rate or progression of sutural Synostosis [2]. Here we present a case of crouzon syndrome in 6 year old boy with review of literature.
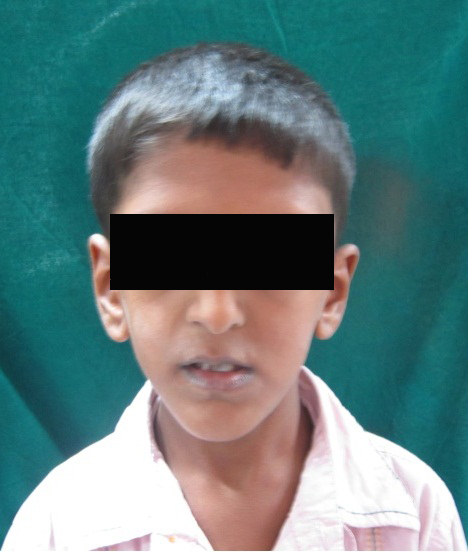
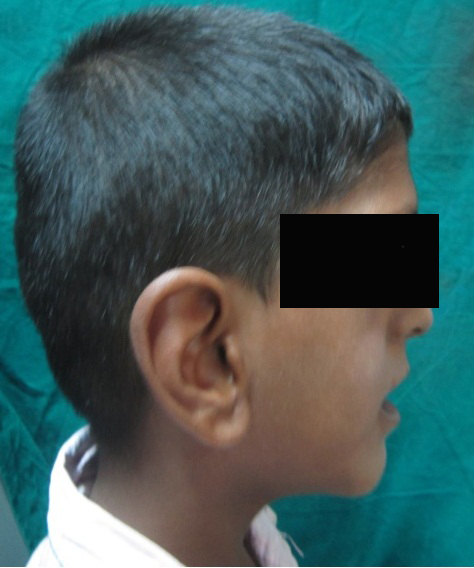
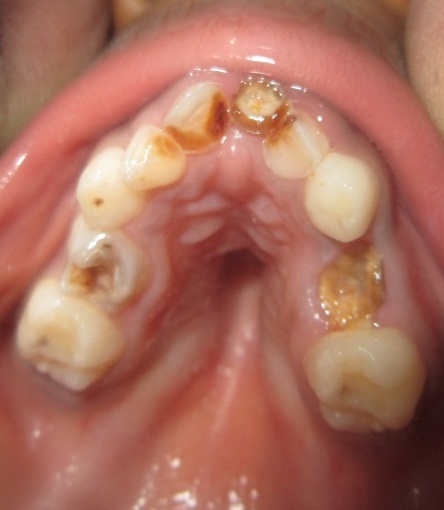
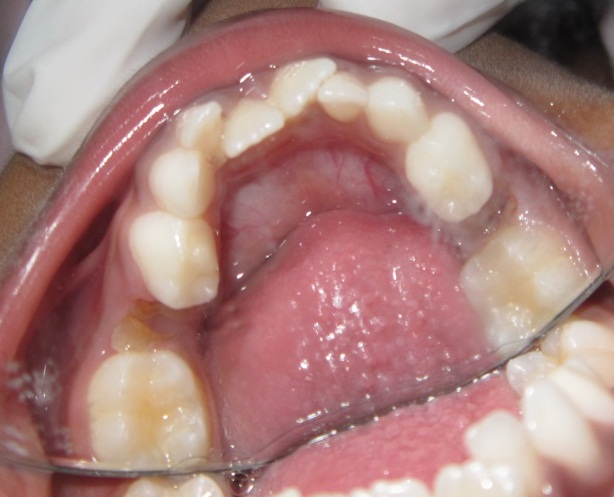
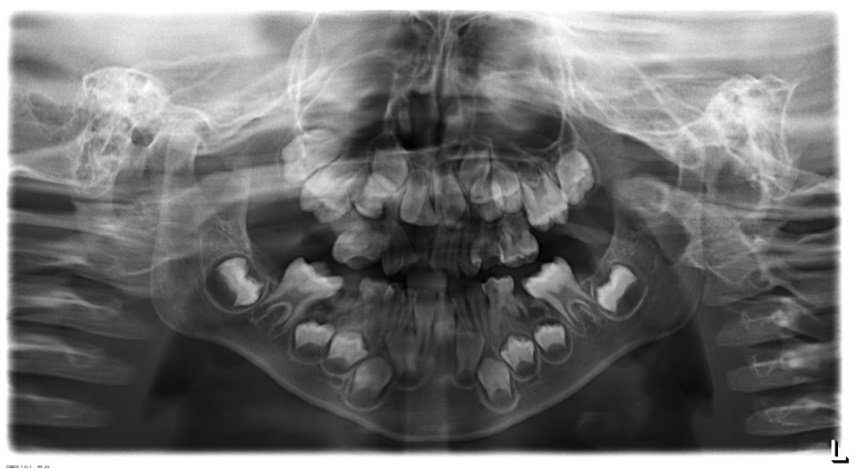
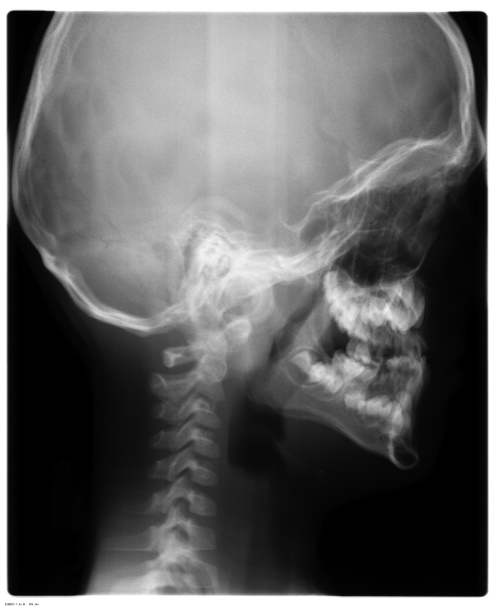
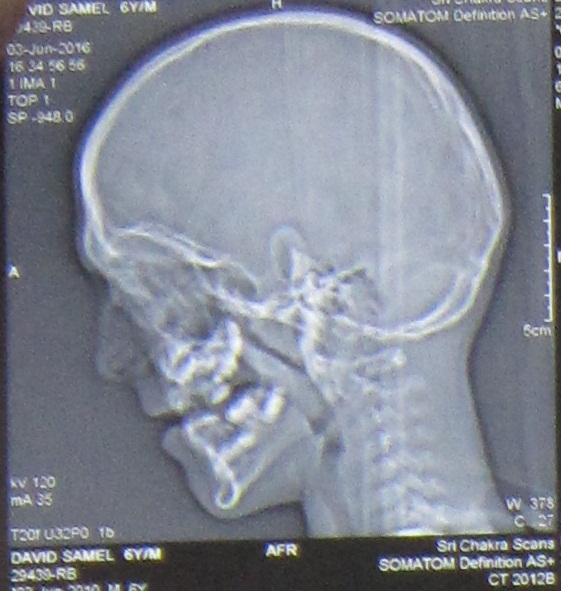
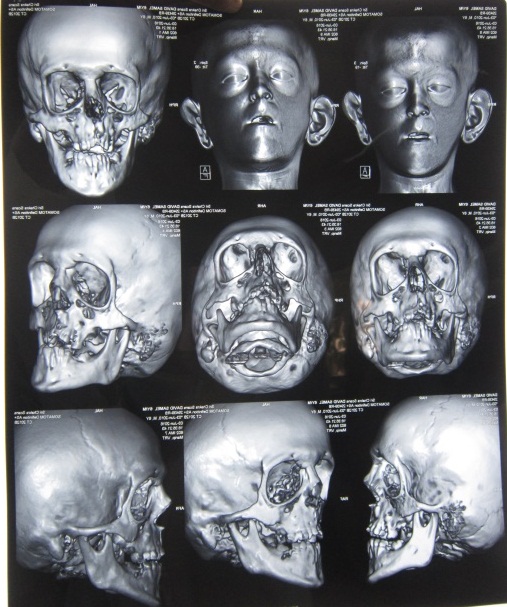
A 6 year old boy (studying 2nd standard) reported to the department of oral medicine and radiology along with his parents with the chief complaint of irregularly arrangement of his teeth for past 1 year. His medical history was non-contributory and his surgical history revealed that the patient underwent surgery for cleft palate at the age of 1 years. The patient presented with obvious dysmorphic cranial and facial features. On extra oral examination the enlarged cranial vault with frontal bossing, maxillary hypoplasia and relative mandibular proganathism was found. Ocular manifestations such as shallow orbits, hypertelorism, bilateral proptosis, and strabismus were present. (Figure 1) Other facial features included short and incompetent upper lip, depressed nasal bridge and enlarged ears without hearing loss. (Figure 2) His hands and feet found to be normal. On intra oral examination, V-shape maxillary arch with high arched palate and bilateral palatal swelling, which was mimicking a pseudo cleft? The number of teeth present was 21, mixed dentition, and multiple decay teeth. (Figure 3 and 4). Patient subjected for radiographic evaluation. Orthopantomogram (OPG) reveals mal occlusion, bilaterally decrease distance between coronoid and condylar and normal trabecular bone pattern (Figure 5). Lateral ceph revelas hypoplastic maxilla, shallow orbit with prominent cranial markings of the inner surface of the cranial vault seen as multiple radiolucencies appearing as depressions resulting in the ‘hammered silver’ (beaten metal/copper beaten) appearance indicating internal remodeling of the calvaria due to an increase in intracranial pressure as a result of premature cranial suture fusion (Figure 6). Computed tomography (CT) reveals deformation of bilateral lateral orbit wall, shortening of right zygomatic arch, bilaterally decrease distance between coronoid and condylar process, V shaped right anterior cranial fossa, mid facial retrusion, high arched palate and malocclusion of the teeth, enlarged adenoids and bilateral tonsils (Figure 7). Other systemic examination was found to be normal. Routine haematological and biochemical tests were within normal limits. With this the final diagnosis of crouzon syndrome was given. The patient is currently under the treatment for malocclusion and all the decay teeth have been restored.
Crouzon’s syndrome was first described by a French neurologist, Octave Crouzon in the year 1912 as one of the varieties of craniosynostosis. It is otherwise called as craniofacial dysostosis [1]. It has a prevelace of 15-16% in one million new born and 4.5% of all craniosynostosis. The genetic defect from mutation in the fibroblast growth factor receptor 2 (FGFR-2) on chromosome locus 10q25q26 which results in the early fusion of the skull bones during the development of the foetus. Cranial malformation in Crouzon syndrome depends on the order and rate of progression of sutural Synostosis [2].
Crouzon syndrome, described as one of the varieties of craniofacial dysostosis, is caused by premature obliteration and ossification of two or more sutures, most often coronal and sagittal. Some authors connect those syndromes as one, calling it Crouzon-Apert syndrome but symptomatologic differentiation makes classification difficult [3].
Although there is considerable individual variation in the appearance of patients with craniofacial dysostosis, the signs are all basically due to early synostosis of the sutures. Facial deformity is observed at birth, followed with time by other features of the syndrome. Craniosynostosis commonly begins during the first year of life and is usually complete by 2–3 years of age [4]. In some cases, craniosynostosis may be evident at birth. In our patient also the features started developing from the birth gradually given by history of the parents.
Occasionally no sutural involvement may be noted. Shallow orbits and ocular proptosis are diagnostic features of Crouzon syndrome. Ocular proptosis, a feature occurring in 100% of the cases, is secondary to shallow orbits and results in a high frequency of exposure conjunctivitis or keratitis. Luxation of the eyeglobes has been observed in some cases. In our patient all the features of craniosynostosis as well as feature of ocular proptosis is seen but ketatitis was not presented.
Low frequency findings include nystagmus, iris coloboma, aniridia, anisocoria, corectopia, microcornea, megalocornea, keratoconus, cataract, ectopia lentis, blue sclera, and glaucoma. In our patient all these feature was not present.
Approximately 50% have lateral palatal swellings but only in a few instances are they large enough to produce the median pseudocleft palate appearance found so frequently in Apert syndrome. In our case the palatal swelling was present which mimics the pseudocleft.
Because of maxillary hypoplasia in Crouzon syndrome, the anteroposterior dimension of the maxillary dental arch is shortened. Dental arch width is also reduced, and the constricted arch gives the appearance of highly arched palate, although palatal height is normal by measurement. Progressive hydrocephalus, chronic tonsillar herniation, and jugular foramen stenosis with venous obstruction occur with significant frequency. Headaches were found in 29% in Kreiborg’s series. Seizures occurred in 12%, and marked mental deficiency was found in only 3%. In our patient there was no such headache, seizures and mental deficiency.
Conductive hearing deficit is found in 55% and atresia of the external auditory canals occurs in 13%. Deviation of the nasal septum was observed in 33% of Kreiborg’s series. Calcification of the stylohyoid ligament was especially common, being found in 88%. Cervical spine anomalies were also common 30% [5]. In our patient, present with enlarged ears with no hearing loss, has a shallow nasal septum without any deviation. There was no calcification of the stylohyoid ligament and cervical spine anomalies. Differential diagnosis of Crouzon syndrome includes Apert syndrome and Pfeiffer syndrome. Apert syndrome will have all the manifestation that will be seen Crouzon syndrome except for the syndactyly of hands and feet. In case of Pfeiffer syndrome broad thumbs, broad big toes and partial and variable soft-tissue syndactyly of the hands and feet will be presented in additional to the features of Crouzon syndrome [6].
A neurosurgical procedure is recommended in cases of intracranial hypertension leading to further optic atrophy. The surgery is difficult, and the procedure must be considered and undertaken in stages. Plastic surgery of the face could be of great help. It is one of the few syndromes where the cosmetic results of the surgery can be strikingly effective [7]. These patients may ultimately come to lead a relatively normal life. In our patient, we plan for orthodontic treatment during the growing stage and a final orthogenetic surgery will be performed after the puberty if required.
Crouzon syndrome is relatively one of the rare syndromes. Dental professionals should have sufficient knowledge of syndromes associated with dysmorphic faces to detect patients who are unaware of their condition, so they may be identified and sent for early investigations and management as required to prevent complications due to late diagnosis.