Identification of a New Site of Metabolism for Phenprocoumon by Modeling it’s CYP2C9 Hydroxylation Pattern
Molecular modeling was conducted to study the observed sites of metabolism for cytochrome P450 enzyme CYP2C9 and find a hitherto unknown hydroxylation site for phenprocoumon. Four hydroxylation sites of phenprocoumon metabolism by CYP2C9 were taken from our previous site of metabolism (SoM) identification by mass-spectrometric and selected-reaction-monitoring chromatographic analyses. Phenprocoumon (PPC) and warfarin (WFN) were successfully docked into CYP2C9 and CYP3A4. Docking results for the four monohydroxylation metabolites (6-OH-PPC, 7-OH-PPC, C2’-OH-PPC and C4’-OH-PPC) showed final docking poses in good keeping with the experimental data. The catalytic cycle was revised and ternary coordination complexes between heme B, reactive oxygen and substrate with a tetrahedral geometry (atoms Fe-O-CH) were identified under the molecular mechanics force field (Yeti). Additional quantum mechanics calculations (Gaussian09) also focused on the ternary coordination complex (iron IV - activated oxygen - sp3 hybrid carbon atom). Transition state and equilibrium structures for OH-Ph-S complexes of iron porphyrin moiety were computed with density functional theory (DFT) at B3LYP level of theory. To establish the most preferred mode of interaction between the complexes and the protein structure, the DFT calculations have been used to accomplish the molecular docking studies. As a direct result the hitherto unknown structure of a PPC metabolite could be identified as the 10-hydroxy-metabolite of PPC. To simulate the hydroxylation of the ortho position (2’) at the phenyl ring, however, a customary model (rotamer library) was generated. After rotation of three binding-relevant side chains (Val113, Leu365 and Thr301) the sterically congested 2’ position became accessible to enzymatic hydroxylation.
Keywords: 4-hydroxycoumarin; Vitamin K; Warfarin; Docking pitfalls: Ab inito
List of abbreviations: PPC: Phenprocoumon; SRM: selected-reaction-monitoring (chromatogram); TS: Transition state; MS: Minimum state; WFN: Warfarin
Acenocoumarol (ACE), phenprocoumon (PPC) and warfarin (WFN) are commonly used for the causal or prophylactic oral treatment of venous and arterial thrombosis (vitamin K–dependent blood clotting). Receivers of oral anticoagulants show a large variability in bleeding risks. Bleeding is a side effect of hospitalized patients on anticoagulation therapy which occurs with a frequency between 15 to 20 percent [1]. Impaired biotransformation and elimination of frail patients may contribute to the variable bleeding risk known as hemorrhagic risk by coumarin anticoagulants. The molecular target of these anticoagulants is the vitamin K epoxide reductase complex [2]. These drugs undergo vast biotransformation in the liver by members of the microsomal metalloenzyme family known as cytochrome P450 oxygenases (CYP for short). Our target enzyme CYP2C9 as well as other members participate in phase 1 biotransformation of body substances or xenobiotics (drugs) by demethylation, oxidation, or hydroxylation reactions [3]. Prior to our computational molecular simulation study, the relative human liver microsome contents of CYP2C9 were assessed against other CYP liver enzymes (CYP1A2, 2A6, 2C19, 3A4) by means of spectrophotometric measurements by a group member [4-8] (Figure 1). In particular five monohydroxylated phenprocoumon metabolites were identified (positions 6, 7, 8 2’ and 4’) in human plasma by liquid chromatography mass-spectrometric coupling [6]. Our present in silico study aims at modeling those five experimentally identified hydroxylation sites of phenprocoumon (figure 2 in [6]). In view of the enzymatic mechanism based on the heme group at the active site of all CYP enzymes and the fact that all monohydroxylated metabolites – except for one – were analytically identified, we also aim at delivering the long-awaited answer to the unsettled question whether the hitherto unidentified peak in the chromatogram could be ascribed to one of the remaining three structural positions at either C9, C10 or C11 (figure 2 in [6]). Thus, the additional goal was to computationally identify the missing hydroxylated PPC metabolite (C9 or C10 or C11-OH-PPC) [4-8].
To our best knowledge substrate coordination to a multi-step catalytic cycle is a nontrivial endeavor. This way, we had also to unveil which catalysis step is amenable to a molecular mechanics formalism (docking algorithms), and whether standard docking tools encompass the atom types of the catalysis configuration (intermediates, transition states) and therein be applicable to substrate - enzyme docking (coordination complex). Another not less worrying challenge was to face the renowned induced fit mechanism since standard docking means coupling a flexible ligand to a rigid receptor. In particular, this induced fit phenomenon occurs upon ligand - receptor complex formation and consists of conformational changes of one or both molecular partners. They can comprise smaller subtle changes in torsional angles at side chains, or even main chain movements on the proteins, and entire domain rotations at a large scale of multi-subunit, multimeric complexes. To further the reader’s interest, three reviews lend detailed insight into this topic of protein flexibilities [9-13].
Molecular docking is the mainstay computational technique to describe the binding between a ligand to its target receptor protein. Each ligand pose is evaluated by a score to reflect its binding affinity [14]. It is, however, not straightforward to use any conventional docking tool to compute the phenprocoumon (PPC) binding to the heme group at the catalytic site which forms a monodentate coordination between the metal center and the substrate intermediate during the oxidative biotransformation step in the liver cells [15].
To this end we combined molecular mechanics (ligand docking to receptor) and quantum mechanics (ab initio transition state calculations of the coordination complex) to generate proper molecular models of the sites of metabolism (SoM) upon PPC binding to the active site of CYP2C9.
Almost all drugs are noncovalently bound to their receptors. But in rare cases a reversible or irreversible covalent bond is formed between them (e.g. inhibitors like carbapeneme of penicillinase or parathion of cholinesterase). Hence, new docking techniques have been evolved for covalent docking (e.g. Covdock, Covglide). In a seminal work by A. Bender and coworkers, the software which can predict CYP activities was reviewed [16]. Moreover, the more general concept of ligand - receptor docking has already been applied to the particular field of substrate - CYP enzyme metabolism (Table 1). Thereby, the most frequently used programs include Autodock 4, AutodockVina, GOLD and FlexX [17-20].
Computing the site of hydroxylation for the structurally undefined OH-PPC metabolite seems a reasonable endeavor since molecular modeling techniques have already been applied to the field of CYP metabolism (figure 2 in [6]).
The complex affinities were assessed by ligand - receptor docking simulations (Sybyl FlexX, Autodock 4 and Yeti 8) [17,20,29]. This process engages the sixth coordination bond, the electron movements of which cannot be directly computed using any molecular mechanics (docking algorithms). Hence, it was treated separately by an ab initio approach (Gaussian software package) [17,30,31].
The crystal structures were downloaded from PDB, their superposition conducted under Swiss PDB Viewer, and manual docking was carried out under VEGA ZZ [32-34]. Automated docking was performed by FlexX (Sybyl X) and Autodock 4 (MGL tools) [17,18,20,35]. The final poses were treated under Yeti 8 and Biograf R [36,29]. We point out, the rationale behind the decision to refine local minima of the final geometries lies in Yeti’s implementation of specific parameters for iron and heme groups. Precisely, it constitutes an extension to the AMBER force field [36]. Related crystal structures of ligand-CYP enzyme complexes (coumarin ligand with PDB code: 1Z10 and flurbiprofen with PDB code: 1R9O) were inspected and back docking of the extracted ligand evaluated to validate the blind docking of PPC into human CYP2A6 or CYP2C9 [37,38].
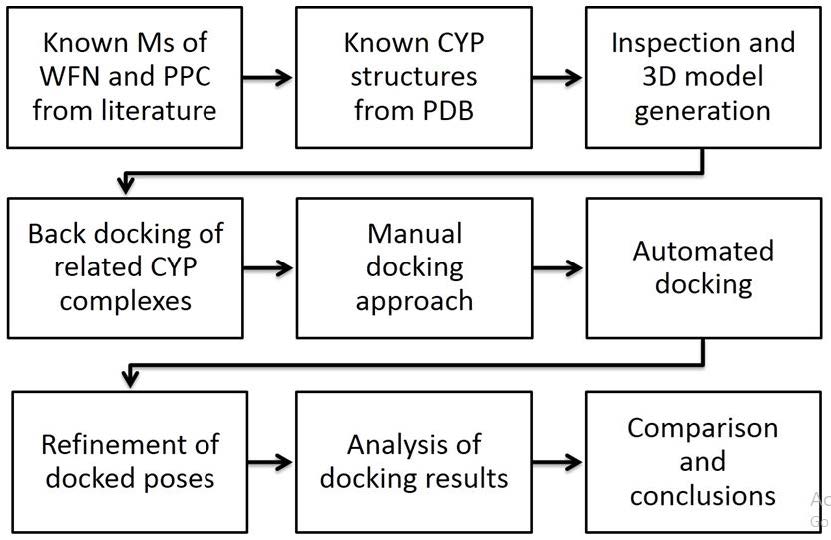
For decades docking tools have focused on the noncovalent interactions, but we cannot neglect the pivotal role of heme at the active site. Between enzyme and its substrate there exist potential energy contributions from - not only noncovalent interaction – but also complex coordination. While the former are built-in components of parameterization sets of conventional docking algorithms, the latter are nonstandard features [17,29]. A schematic workflow of nine steps was devised to combine manually docked poses as start positions for automated docking by Autodock and refinement of final poses by Yeti 8 (Figure 1) [17,29].
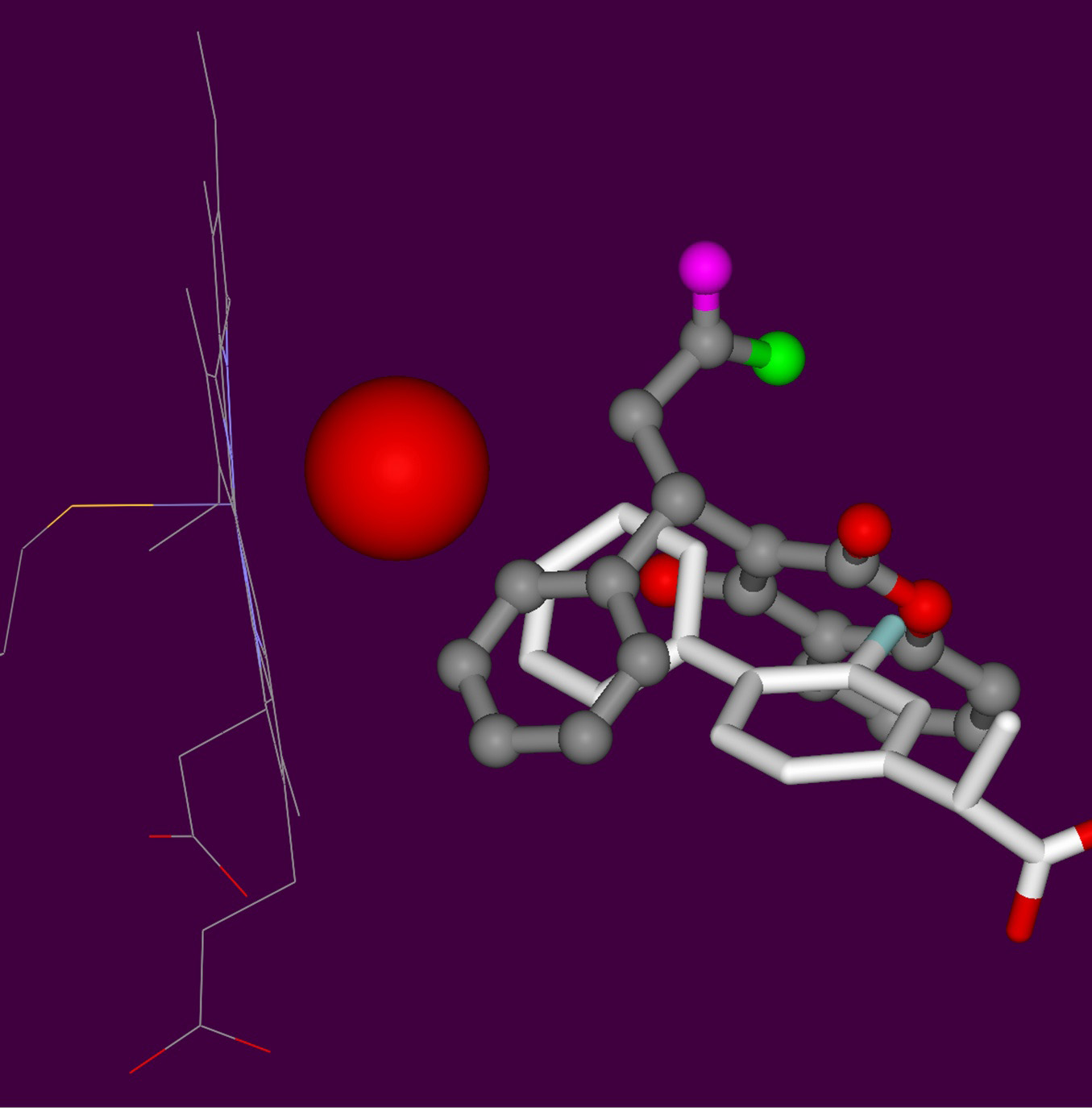
Mass spectroscopic evidence provides a strong indication that the phenyl ring of the substrate is hydroxylated at the 2’ (two prime) position. The final docking poses showed a ligand orientation similar to the crystal structure of flurbiprofen with significant resemblance to structural features of PPC (flurbiprofen, PDB code: 1R9O) [6,8,38].
After comparing their structures we carried out selfdocking to assess which step of the catalytic cycle is suited as the final pose (docking endpoint) and how to coin this substrate - enzyme contact into the molecular mechanics formalism for Autodock 4 and Yeti 8 (Figure 2).
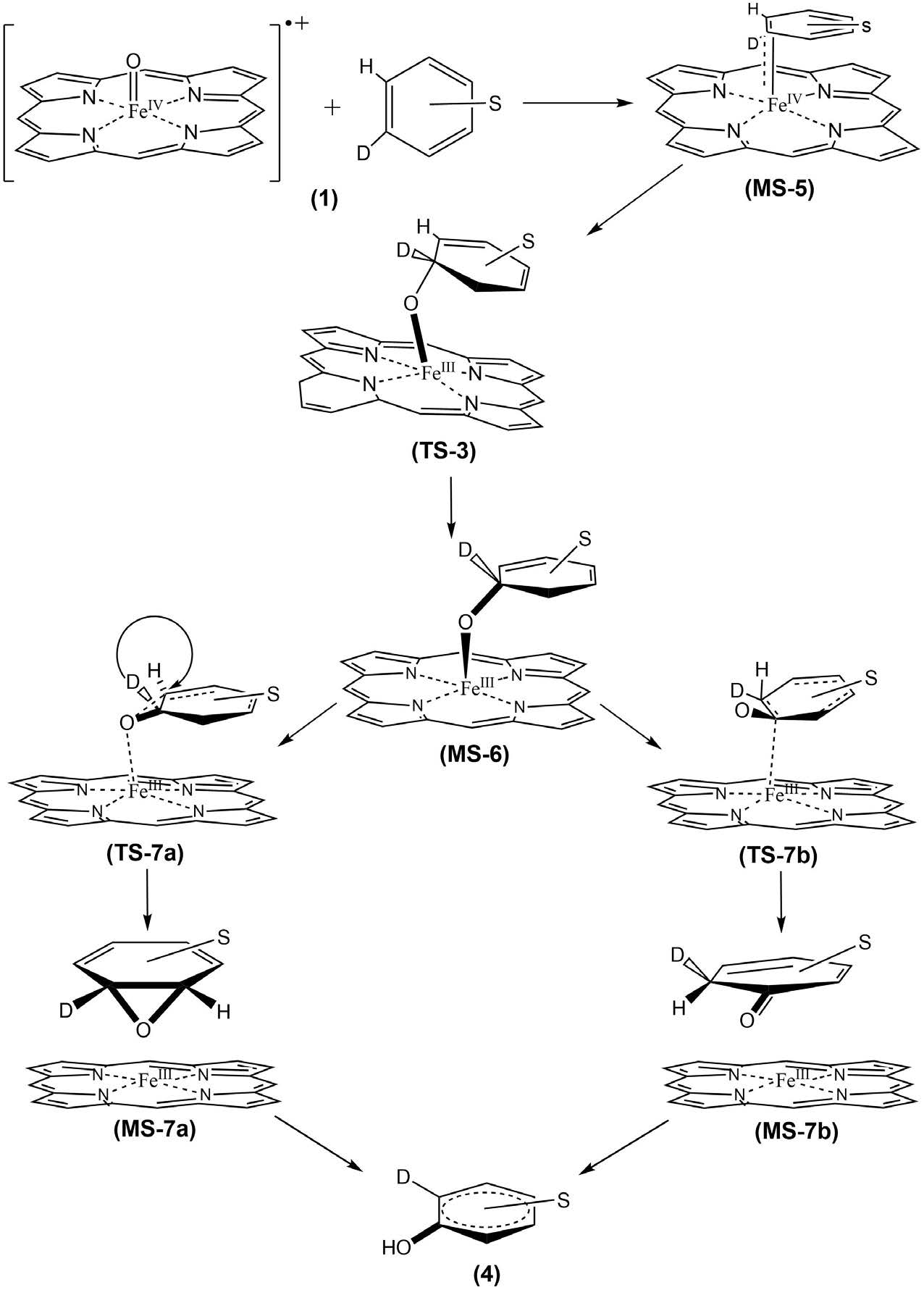
In this work, a theoretical study of the mechanism of reaction between the heme group and the aromatic ring is presented, using DFT-B3LYP method. A model of the general mechanism is proposed for this reaction in gas phase (Figure 3).
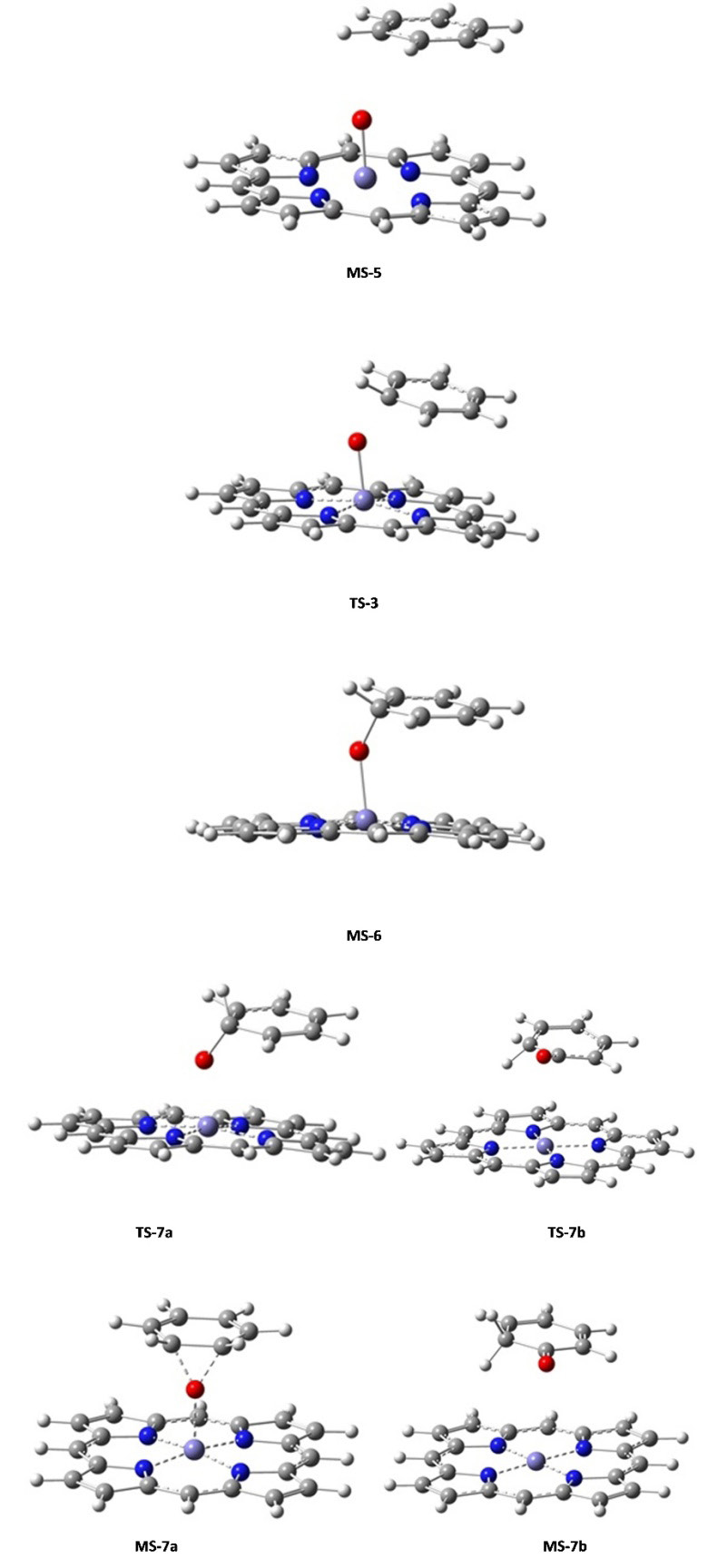
In the first step, from (MS-5) structure, it is proposed the formation of a tetrahedral transition state (TS-3) for obtaining the intermediary (MS-6). This intermediary has the structure like a ternary radical cationic complex formed by a central carbon atom with temporary sp3 hybridization: a type of reactive oxygen and the cation of iron as a catalyst with charge +III (FeIII) on the side of heme group. The second step of the reaction is the posible formation of two species (MS-7a and MS-7b): one corresponds to the formation of an epoxide group and another of a keto group, both from their respective transition state structures (TS-7a and TS-7b). These last species are completely dominated by electronic influence in order to stabilize the corresponding intermediaries (MS-7a and MS-7b). Finally, in the case where the alcohol is only involved as final product (4), an hydrogen atom was replaced by deuterium (D) to analyze the mechanism known as “NIH-shift”, whose final product is of interest in this study for molecular docking simulation [39].
Regardless of the subtitle (quantum) differences between both reaction coordinates, the aforementioned tetrahedral ternary complex constitutes the point of departure in common for both. Hence, that configuration is chosen as a static end point of docking and as a predefined transition state (TS) for ab initio calculations.
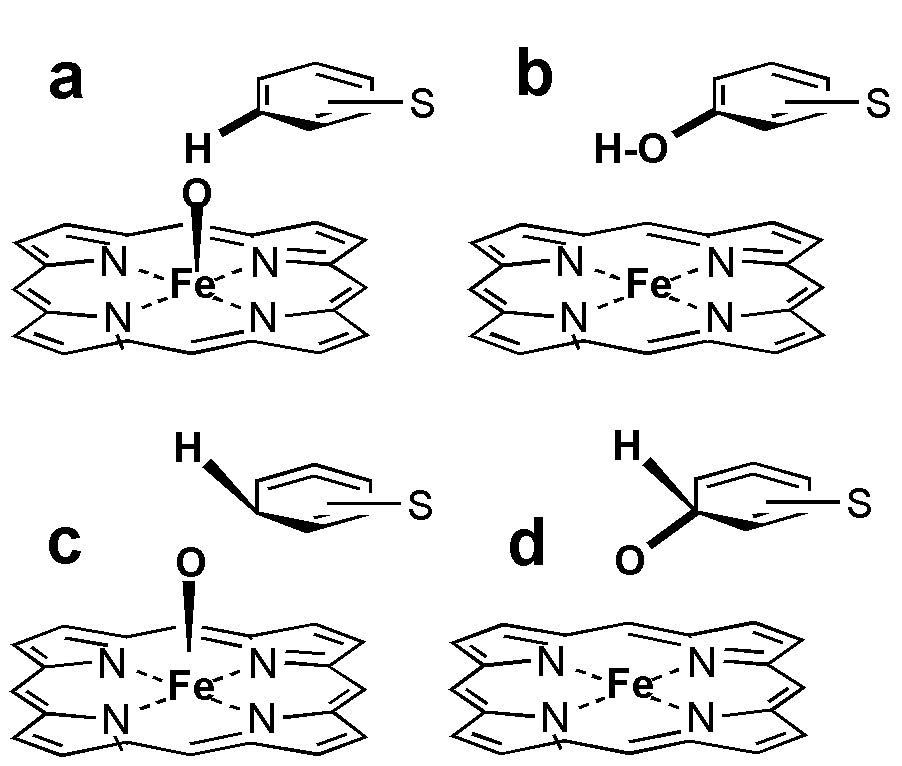
Best ranked docking solutions were extracted from the 2.5 Å RMSD clusters (stage 6) for binding energy refinement (stage 7). At stage 7 (Figure 1) the docked ligand complexes were refined under the Yeti force field. It is an AMBER extension and includes parameters for ion and heme [29]. Thereby, all final poses obtained a natural octahedral configuration and were ranked under Yeti 8 [29,40]. At stage 8, inspection and evaluation of the final poses took place. The accepted docking solutions were compared to the binding geometries observed in the crystal structures of hydroxylated camphor CYP P450 complex [41,42]. The x-ray structure of CYP2A6 with co-crystallized coumarin was used as another 3D-template because it revealed the geometries between substrate and the heme - oxygen system (PDB codes: 1Z10, 2CPP and 3CPP) [41,42,37]. Coordination geometries were measured: Fe-O-C atom distances, Fe-O-C angle, and dihedral angles between the heme group and docked ligands (Figure 4) [43].
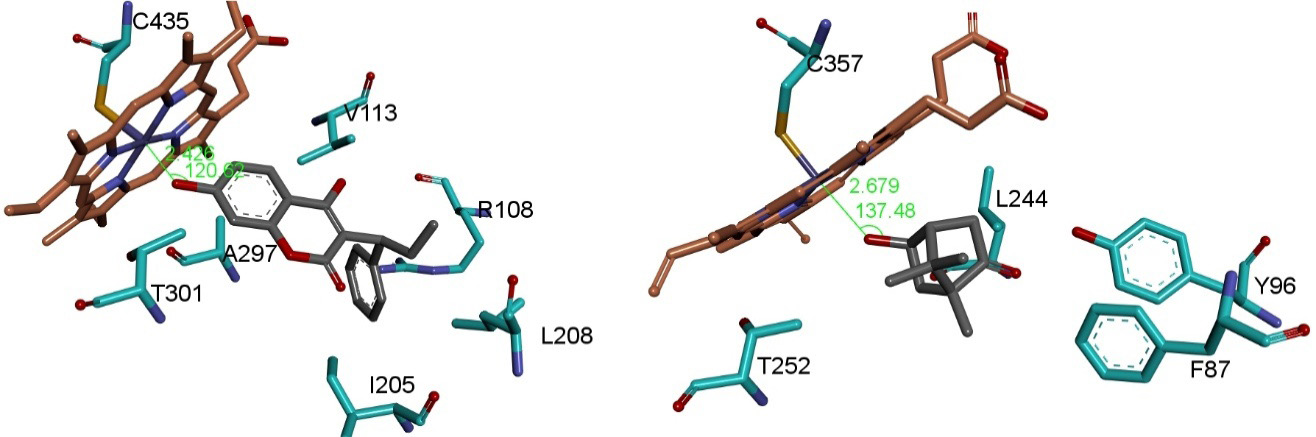
No crystal complex with PPC exists, but with structurally related WFN which was co-crystallized with a mutant type CYP2C9 (PDB code: 1OG5) [44,45]. So we decided to study WFN, too. With the catalytic site nearby the mutated side chains, steric hindrance caused the substrate displacement further away from the heme group. Finally, the refined poses from Yeti were found plausible upon comparison with related crystal complexes (Figure 5) [38,41,42,44-47].
In the case of PPC C2’ we found indications for induced fit with docked solutions reflecting conformations other than the available crystal structures (Figure 5). At stage 9, we compared the observed chromatographic data to the calculated affinities for correlation between the amount (AUC) and the potential energies of docked poses (Table 2) (figure 2 in [6]). Upon analyzing the binding energies of the final poses it can be seen to illustrate one instance that C7 is energetically preferred over C6 hydroxylation. This is in good keeping with the SRM chromatogram (figure 2 in [6]).
The hydroxylation pattern of the main metabolites (see circle 1 = 4´-OH-PPC; circle 2 = 2´-OH PPC; circle 4 = 6-OH-PPC; circle 5 = 7-OH-PPC ) were successfully reproduced except for circle 2 due to steric hindrance. This is a hint for shifting side chains or backbone (induced fit) which cannot be handled adequately by rigid protein docking procedures (Figure 2) (Table 2) [6]. As source of the steric clashes we identified Val113, Leu365 and Thr301 on CYP2C9 (PDB code: 1R9O, resolution 2 Å, R value 0.2) [38]. To avoid bad substrate - enzyme contacts (clashes) new conformations for their side chains were selected from a rotamer library to allow the 2’ hydroxylation [33,49].
The spectral data could also be reproduced in a semi-quantitative way: the (S)-enantiomer always fitted better into the catalytic site which explains why the (S)-metabolites always appears under a smaller area under curve (AUC) than the (R)-metabolites of PPC. The biotransformation rate of the (S)-forms should then be higher to explain why the blood (plasma) concentration was smaller. This parallels the finding that CYP2C9 is the main metabolizer of S-WFN but only marginally of R-WFN substrate [6,50].
Yet these findings leave one question still to be answered concerning the peak labeled as circle 3 in the spectral analysis (figure 2 in [6]). To complicate matter, early attempts in docking PPC for the ortho-position (2’) on its phenyl ring fell short of expectation. Probing with WFN it was found that the ortho-position (2’) on the phenyl ring actually was accessible to oxygenation (Table 2). The apparent contradiction was resolved acknowledging that PPC was docked into the empty active site of the crystal structure liganded to flurbiprofen [38]. Apparently the ligands invoke induced fit phenomena leading to structural rearrangements between PPC substrate and enzyme (spatial requirements). Since no crystal structure of PPC - CYP2C9 has been elucidated, we simulated the missing induced fit by rotamer changes (Val113, Leu365 and Thr301). This way C2’ hydroxylation of PPC was successful. This finding extends the known pattern of metabolism which we summarized in the introduction chapter. Playing back the final poses for PPC and WFN, position C9 on both were clearly an unlikely place for hydroxylation because the C9 ramification lead to spatial congestion shielding against catalytic attack (Table 2).
The understanding of steric requirements as the essential driving forces for regioselectivities was furthered by two facts from literature: (i) C9 is not metabolized on WFN and (ii) the more accessible position C10 of WFN is hydroxylated instead. In good keeping the C10 hydroxylation of PPC was the most populated solution cluster after scoring the docked poses (Table 2). On theoretical grounds it was concluded without ambiguity that the peak without structural information labeled as circle 3 be ascribed to C10-OH-PPC. C10-hydroxy-WFN was found to be the second most abundant plasma metabolite (Figure 2) [51].
Our ab initio study was carried out to generate possible transition states by quantum mechanics.
Albeit, it does not lie within the scope of this study to evaluate the extant literature on heme enzyme docking it is save to utter that this study explored all four options to formulate the docking start geometries to reflect the catalytic circle on both, a molecular mechanics level defining adecuate atom types and on quantum mechanics level, too (Figure 2).
In order to analyze the electronic and geometrical propensities used as starting geometries of the molecules for docking studies, the reaction mechanics was studied by a quantum mechanics approach. The structures of reactants, transition states and products were optimized. To this end only functional parts of the larger molecular entities were used, so that the fragments were amenable to “ab initio” treatments (Figure 6 and 7).
A hybrid-GGA functional, named B3LYP, was used for carrying out the calculations. Hybrid functionals mix exchange energies calculated in an exact (Hartree-Fock-like) approach with those obtained from DFT methods in order to improve the task performance. Several analyses on the assessment of the B3LYP performance have been made for interactions in bio-molecules and clusters which confirm the accuracy of this method [52-54].
The optimization for the present quantum mechanical study was performed at the B3LYP level using the 6-31G basis set applied to C, H, O and N atoms [55]. A relativistic effective core potential (ECP) on Fe atom was used to replace the inner core electrons, keeping the outer core electrons. In addition, the harmonic frequency calculations and zero-point vibrational energies (ZPE) were calculated at 298.15 K without considering any scale factor using the same theory level [56]. The free energies (G) of the reactants, intermediates, transition states, and products were obtained using the equation G = Etotal + ZPE − TS, where Etotal is the total energy of the species, T is the temperature in Kelvin and S is the entropy. All calculations were carried out using the Gaussian09 program [30]. The optimized geometries structures were visualized using GaussView 5.0.8 graphical application [57].
The structure and electronic energy of iron-porphyrin (FeP) system were calculated in gas phase. Optimized geometries for three oxidation states (Fe(II)), Fe(III), and Fe(IV)) and different multiplicities for iron were evaluated (Table 3). According to the obtained results, the most stable specie corresponds to 3Fe(II). The following trend in relative energies, in kcal mol-1, is observed from this structure: 1Fe(II)< 5Fe(II)< 4Fe(III)< 2Fe(III)< 6Fe(III)< 5Fe(IV)< 1Fe(IV)< 3Fe(IV). A value of 12 kcal mol-1 between the most stable specie and 1Fe(II)is found, while concerning 3Fe(IV) the relative energy is of 737.75 kcal mol-1.
An important role in the applications of quantum chemistry calculations for molecular systems is the calculation of Mulliken charges, because the charge on atoms affect to properties as electronic structure, molecular polarizability and dipole moment, among others. For example, the distributions of charge on atoms forming molecules suggest the formation of acceptor or donor pairs involving charge transfer. In addition, the atomic charge analysis can be used for describing electronegativity and it is also useful for charge transfer processes in chemical reactions. The results of Mulliken charge densities were also calculated using the B3LYP method.
An interesting correlation between the charge and the interatomic bond Fe_Np was found, being Np the nitrogens of the porphyrin ring. When a major charge exists in the system, a decreasing interatomic bond Fe_Np is obtained. The Mulliken atomic charges show, in all the cases, that the iron atom has a positive charge, while the charge on nitrogen atom is negative. These results demonstrate that when the iron atom changes of FeII to FeIV (major charge in the system) the charge is major on iron atom. So, the nitrogen atom has a value of maximum negative charge close to -0.710 in the quintuplet state of FeIIP. The maximum positive charge is obtained for the Fe in the quintuplet state of FeIVP, whose distance Fe_Np corresponds to the small value found, showing the charge-distance relationship for the different systems studied in this work.
As a most valuable asset, our theoretical results obtained here are in good agreement with experimental solid state results and previous theoretical calculations obtained by BLYP/DPN level of theory [57,58].
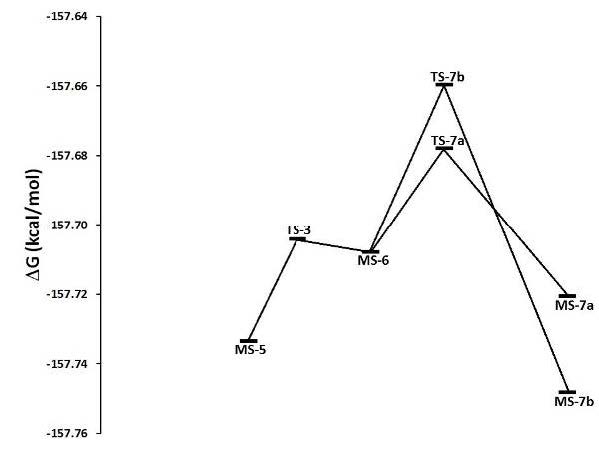
Concerning the reaction mechanism the potential energy surface (PES) profiles assessed the energy minima, intermediary and transition state geometries. Three main transition states (TS-3, TS-7a and TS-7b) were found for the second stage of the hydroxylation mechanism (Figure 3). At that stage, the benzene moiety started its approach and the iron complex formed an intermediate (Figure 7). The transition state called TS-3 then showed an activation free energy of -157.7042 kcal mol-1 which fairly lay above the value of intermediate MS-5. Then the complex iron-porphyrin-O-phenyl (MS-6) was formed with a free energy of -157.7080 kcal mol-1 (Figures 3 and 6). In this step, about -0.0038 kcal mol-1 was delivered upon complex formation, all of which reflected the feasibility to form this complex as an intermediate (Figure 7). Prior to our work, two ways toward the production of the final compound had been experimentally observed (Figure 7) [39]. Both were amenable to model simulation during the third step: the migration of the Deuterium atom (atom D replacing atom H to observe the NIH shift) between two carbon atoms in both cases allows the complex formations MS-7a as well as MS-7b. The migration occurs through the TS-7b formation, with an activation free energy of -157.6596 kcal mol-1 at the expense of -0.0484 kcal mol-1. Once migration has been achieved, the final alcohol product can be obtained from TS-7a, with an activation free energy of -0.019 kcal mol-1 what fairly lies below the energy level of TS-7b with an energy consumption of -0.0299 kcal mol-1. For this last step of product formation, a mechanistically plausible intermediate (MS-7a) was calculated and its formation energy below the intermediate (MS-5) was assessed: -157.7206 kcal mol-1 which reflects the energy release of 0.0887 kcal mol-1. All values of formation and activation free energies were calculated and compared relative to the ground state (Table 3). The respective geometries are taken into account to formulate the discrete geometries (Figure 7). The Figure 7 shows the minima and transition state structures. Three main transition states (TS-3, TS-7a and TS-7b) were found in the second step of the mechanism. In this step, the biomolecular process begins with the approach of the benzene molecule and the complex FePO, forming the intermediate (MS-5). The transition state TS-3 (Figure 6) showed an activation free energy of -157.7042 kcal mol-1, while the complex iron-porphirin-O-phenyl (MS-6 in Figure 6) was modeled to assess the free energy of -157 kcal.mol-1. The formation of the complex MS-7b is obtained from the transition state TS-7b, with an activation free energy of -157.6596 kcal.mol-1. On the other hand, the alcohol formation can be obtained from the TS-7a, with an activation free energy of -0.019 kcal.mol-1 below TS-7b (Figure 3). In this last process, a possible intermediate (MS-7a) is calculated, with a formation energy of -157.7206 kcal mol-1. The values of formation and activation free energies are shown in Table 4. The respective geometries are depicted in (Figure 6 and 7). The discrete geometries are displayed in Figure 7.
The calculated structural parameters of the complexes are presented in Table 5. The average of the interatomic bonds Fe_Np in the complexes is of 2.01 Å, while the interatomic distance Fe-O for the two complexes TS-7b and MS-7b are 4.18 and 3.54 Å, respectively. A major changes was observed for the distance Fe-O in the acetone formation respect to the alcohol formation. This computed finding reflects probability/feasibility for the complex formation concerning the keto group during reaction. These ab initio geometries are in line with the preselected force field geometries with built-in atom types (Figure 2).
Prior to docking, the identification of adequate geometries was required which were amenable to molecular mechanics formalisms dictated by predefined bond and atom types, their geometries, forces and charges without electron movements but electrostatics. Our proposed prerequisite might not always have been fulfilled in the literature (Table 2). Many authors do not even mention whether the reactive oxygen moiety was present or not, not to mention its attachment to the iron or substrate. In the present case of monoxygenase metabolism, the tetrahedral geometry of the substrate intermediate at the site of (mono)oxygenation was generated by an sp3 hybrid state to reflect the activated state of the aliphatic (C10-OH-PPC) or aromatic (6-OH-PPC, 7-OH-PPC, C2’-OH-PPC and C4’-OH-PPC) carbon atoms under catalytic attack by an oxygen moiety and iron IV-monoxygen radical cation.
Molecular mechanics approach with predefined atom types and electron configurations – as well as quantum mechanics with elaborated and flexible electron descriptions – reflected the experimental data about substrate- or region-selectivities of selected CYP family members. As a silently made assumption, however, it has been assumed that the outer vestibule of CYP proteins with the nearby surface of endoplasmic reticulum membranes would not contribute to regioselectivity. Another limitation was found in the warfarin-CYP2C9 crystal complex where the ligand was not directly bound at the catalytic site, but displaced. The enzyme does not show the wild type but a mutation type. Hence the displacement of ligand was ascribed to those mutated amino acids and the ligand position qualified as an artifact [45].
In the present docking study, Autodock poses were refined by Yeti and both were validated by selfdocking hitherto known liganded crystal complexes of enzymes. PPC and WFN as ligands were successfully docked into CYP2C9 and CYP3A4. Docking lend insight about the steric nature of the driving forces for substrate recognition. The docked poses simulated a ternary coordination complex (iron (IV) - activated oxygen - sp3 hybrid carbon atom). The docking results could reproduce the experimentally determined hydroxylation metabolites of PPC. The structures of four metabolites had already been identified by peaks in mass spectrometry (6-OH-PPC, 7-OH-PPC, C2’-OH-PPC and C4’-OH-PPC).The hitherto unknown structure of the last peak was computationally determined without ambiguity: it constitutes the PPC metabolite with hydroxylation at C10 position (acronym: 10-OH-PPC). Steric clashes for one of the five metabolites (C2’-OH-PPC) reflected induced fit which was simulated using an empiric rotamer library to find observed side chain conformations for Val113, Leu365 and Thr301. In addition, subtle geometrical and electronic movements were calculated by quantum mechanics to describe the reaction coordinate with the transition state geometry of the ligand-heme complex. Our results are all in line with reported regioselectivities. Although it would be farfetched to generalize on the theme, the technical achievement for the field of molecular modeling the enzymatic substrate metabolism becomes obvious: ligand - receptor docking algorithms (advanced force field calculations) can reproduce substrate binding to target enzyme despite the multiple step catalytic cycle.
Graduate student Israel Quiroga is grateful for the Conacyt Grant 360609 during his gradute studies for Master in Science degree during 2013 to 2015. We feel very much beholden to CA-120, CA-263, Faculty Dean Jorge Cerna and VIEP at BUAP for support, and Prof Dr Enrique Meléndez, University of Puerto Rico, Department of Chemistry, PO Box 9019, Mayagüez, PR 00681, USA, for discussion. We thank Dr. David Domeyer and Prof Dr Stefan Laufer, University of Tübingen for FlexX and Sybyl X licening. The authors thankfully acknowledge computer resources, technical expertise and support provided by Laboratorio Nacional de Supercómputo del Sureste de México, which is a member of the CONACYT network of national laboratories. Thanks to Faculty Dean J. Cerna for financial support of publication fees.
BK provided the experimental data about PPC metabolites, FMB provided the ab initio calculations of the transition states, KSA, IQM and TS carried out docking runs. TS created the study design, embedded the literature and prepared the Manuscript.