Identification of the Hub Genes: Case Report
Background:CSystemic inflammatory response syndrome (SIRS) is a primary health concern that needs to be addressed urgently, however, the pathological mechanism of SIRS in genetic level is unclear.The aim of this study was to detect the hub genes and molecular pathways involved in SIRS by using bioinformatics analysis, which may enhance the sensitivity of potential therapeutic signs and diagnostic biomarkers during the process of SIRS.
Methods:The Gene Expression Omnibus database was used to investigate the expression profile of GSE12370. The differentially expressed genes (DEGs) in patients with SIRS and controls were respectively analyzed via limma R/Bioconductor software package and clusterProfiler package in R. The protein-protein interaction (PPI) network data of DEGs, constructed by the Search Tool for the Retrieval of Interacting Proteins (STRING) database, was analyzed by using Molecular Complex Detection (MCODE) plugin of Cytoscape software.
Results:188 DEGs were detected in SIRS samples and controls, including 98 up-regulated and 90 down-regulated genes. Antigen processing and presentation signaling pathways were closely relevant to up-regulated DEGs, and the down-regulated genes controlled the expression of Toll-like receptor signaling pathway. Further, 85 nodes and 278 edges created a PPI network. Molecular Complex Detection analysis was used to screen out the top two important clusters from the PPI. 22 hub genes, including Myxovirus resistance 1 (MX1), MX2, oligoadenylate synthetase-like, interferon regulatory factor 9 (IRF9), IRF1, and IRF8, el at, were selected because of the high connectivity in the PPI network.
Conclusions:The results based on the bioinformatics network analysis identified molecular mechanisms and the main hub genes might accelerate the process of SIRS.
Keywords:Gene Expression Profiling; Hub Genes; Molecular Mechanisms; SIRS
Systemic inflammatory response syndrome (SIRS), an inflammatory state which can affect all systems of the body, is defined as a derangement in routinely observed physiological parameters. It is a nonspecific clinical state,usually diagnosed by the criteria that declared the by American Society of Thoracic Physicians and Society of Critical Care Medicine.SIRS is a common complication after surgery, which can result in significant mortality. Recent examinations detected that the occurrence of SIRS in patients undergoing abdominal surgery ranged between 16% and 89% [1,2]. SIRS was associated with a 13-fold increase in mortality, which could be as high as 4.8%in the aneurysm repair surgeries [3].
The occurrence of SIRS in patients with other diseases, such as subarachnoid hemorrhage [4], alcoholic hepatitis [5] and acute liver failure [6], and even the major determinant of multiple-organ failure, is associated with higher mortality and morbidity rates. However, limited attention to the damage to the immune system, the misuse and overuse of antibiotics, and lack of specificity in diagnosis can increase the morbidity of SIRS. No drugs are currently licensed for treating SIRS. On progression, the disease results in sepsis and septic shock, finally leading to multiple-organ failure and hence resulting in higher mortality [7]. Hence, SIRS is a primary health concern that needs to be addressed urgently.
During the SIRS, large qualities proinflammatory cytokines are released into circulation. The downstream signaling or cytokine cascades are the main responses of SIRS and usually are activated by cytokines which are irritated through the pattern recognition receptors, pathogen-associated molecular patterns, and damage-associated molecular patterns [8,9].However, the physiology of inflammation and the pathogenesis of SIRS are highly complex and not fully understood in many aspects, especially regulatory and molecular mechanisms [10].
Gene micrograms analysis at the level of bioinformatics and network are considerably important to make a thorough inquiry of gene expression profile in disease pathogenesis. During the study,we screened out some hub genes and molecular pathways and tried to identify molecular mechanisms and the main hub genes might accelerate the process of SIRS through different software. The differentially expressed genes (DEGs) between patients with SIRS and controls were compared by bioinformatics analysis. The protein–protein interaction (PPI) network, constructed using Gene Ontology (GO) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis, was operated by the Molecular Complex Detection (MCODE) software [11] .
Microarray data
The Gene Expression Omnibus database (www.ncbi.nlm.nih.gov/geo/), storing curated gene expression datasets, was used in this research.The GSE12370 expression profile data were downloaded from this database. Patients with SIRS after surgery were included in the “SIRS” group,while the “control” group consisted of presurgical patients.A total of 19 SIRS samples and 20 control samples constituted the database. Blood was collected in tubes using venipuncture in the group of control and using a central venous catheter for patients with SIRS. The Affymetrix Human Genome U133 Plus 2.0 Array was used in this study.
Identification of differentially expressed genes (DEGs)
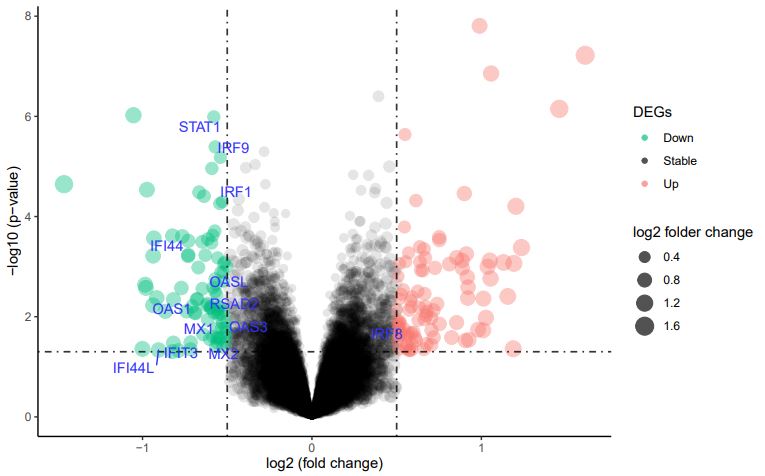
The limma R/Bioconductor software package provides a complete analysis scheme of gene expression experimental data, which can be used for both differential expression (DE) and differential splicing analyses of RNA sequencing [12,13]. It includes importing data, pre-processing (submit reads-per-kilobase-per-million (RPKM) to limma), assessing quality, and normalizing, through to linear modeling, DE, and gene signature analyses during the analysis of gene expression14.And it was used to identify DEGs in SIRS and control in R (Version 3.5.3) [14]. The cutoff criteria were |logFC| ≥2 (log2 fold change) and a P-value <0.05. At last, we found out 188 DEGs which including 98 up-regulated genes and 90 down-regulated genes.
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of DEGs
GO, a classification system through which genes are classified in view of their functional characteristics (including the cellular component, molecular function, and biological process (BP)),was frequently used to comment on the correspondence between gene and GO term [15]. During the examination, we focused on the enrichment results of BP.Also, attempts, from a larger biological view, tried their best to demonstrate the pathophysiological process of SIRS. KEGG pathway analysis, a subclass of KEGG, is always used to demonstrate enriched signaling pathways, which were screened by mapping DEGs to the KEGG database. GO and KEGG enrichment were used to analyze DEGs by the cluster profiler16. It was statistically significant when the P-value <0.05.
Protein–protein interaction (PPI) network and subcluster analysis
The PPI network was always constructed online using the Search Tool for the Retrieval of Interacting Genes (STRING, https:// string-db.org/).The cutoff value was defined as the minimum required interaction score ≥0.4. MCODE was applied to identify the molecular network for module identification according to the clustering of genes in the network.In line with the specific parameters, the MCODE through the vertex weighting by local neighborhood density and outward traversal from a locally dense seed protein could isolate the dense regions [11]. The subclusters in the PPI network of SIRS were created by the MCODE Cytoscape software plugin (Version 3.7.1) [11,17]. We set “degree cutoff = 2; node score cutoff = 0.2; and K-Core = 2”[11]as the selection criteria.
Identification of differentially expressed genes (DEGs)
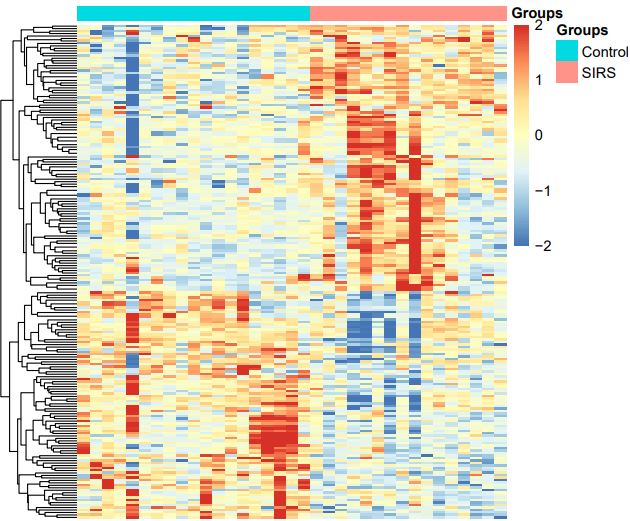
19 patients with SIRS and 20 controls were examined in this study. The limma R/Bioconductor software package was for preliminarily processing the dataset of expression and then we got a heatmap made by top 100 up-regulated and down-regulated genes, showing in the Figure 1.After setting threshold of P-value and logFC, we screened out 98 up-regulated and 90 down-regulated DEGs (Figure 2), which were listed in Table 1.
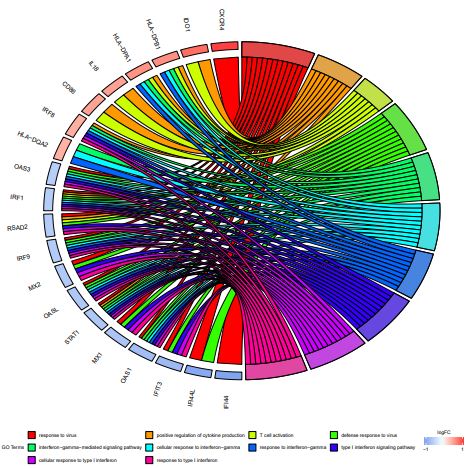
According to results of GO enrichment analysis in BP, the positive regulation of cytokine production, T-cell activation, defense response to the virus, and interferon-gamma−mediated signaling pathway were significantly promoted by the up-regulated DEGs. The cellular response to interferon-gamma,type I interferon signaling pathway and cellular response to type I interferon were closely related to the down-regulated DEGs (Table 2). Antigen processing and presentation signaling pathway were significantly activated by the up-regulated DEGs on the basis of KEGG enrichment analysis. The down-regulated DEGs had a significant influence in the signaling pathway of the Toll-like receptor which were listed in Table 3.
Protein–protein interaction (PPI) network and subcluster analysis
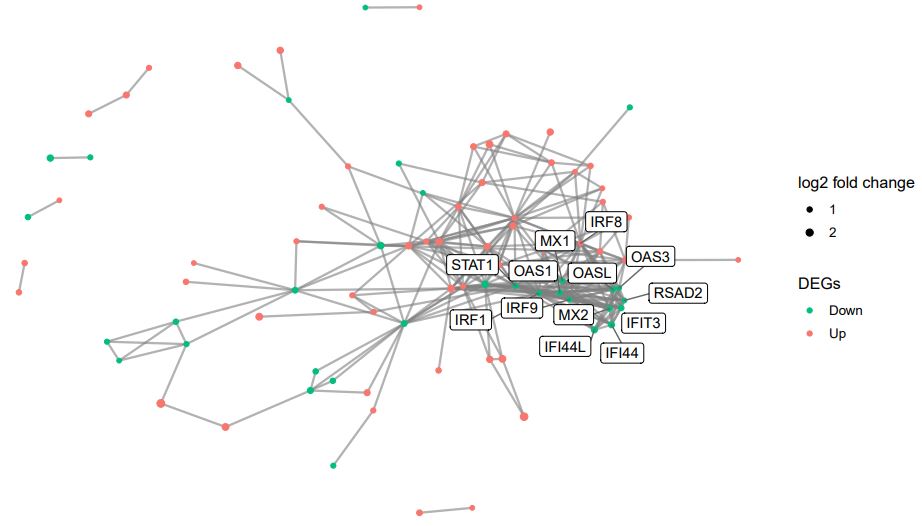
There were 278 interaction pairs of DEGs that were identified from the STRING database, in order to further identify the interaction between genes we constructed a PPI network, including 85 nodes (Figure 3). The subclusters of PPI networks were created via the MCODE Cytoscape software plugin (Figure. 4). Hub genes were defined as those with subclusters having the MCODE score >5. Ultimately, 22 genes were screened out of the 2 subclusters. The GOplot package was used to process the hub genes of the top two subclusters which including the top five GO enrichment analysis terms(Figure. 5)[18].
These hub genes included Myxovirus resistance 1 (MX1), MX2, oligoadenylate synthetase-like (OASL), interferon regulatory factor 9 (IRF9), IRF1, IRF8, 2’–5’-oligoadenylate synthetase 1 (OAS1), OAS3, interferon-induced protein 44L (IFI44L), IFI44, C-X-C chemokine receptor type 4 (CXCR4), CD86, interleukin-18 (IL-18), and indoleamine 2,3-dioxygenase-1 (IDO1), etc. The excessively proliferated and activated of immune cells were certificated closely related to the most up-regulated genes and GO terms, while data showed that the down-regulated hub genes and terms were tightly connected with the influence of the type I interferon (IFN).
The SIRS is an inflammatory state occurring in up to 30% of the hospitalized patients and usually occurs in surgical and intensive care patients [19]. The degree of physical injury, exposure to commensal bacteria, and a whole host of patient factors, including age, preoperative health, and medication, are related to the incidence of SIRS. According to recent studies, the occurrence of SIRS in patients with other diseases, such as subarachnoid hemorrhage [4], alcoholic hepatitis [5] and acute liver failure [6], and even the major determinant of multiple-organ failure, is associated with higher mortality and morbidity rates.A clinical study also demonstrated that the happence of SIRS in oral and maxillofacial surgeries was 32.5%, and SIRS was the pioneering performance of sepsis, which maight to lead multiple-organ dysfunction syndrome[20].It should keep vigilant if the patient occurred SIRS promptly postoperative, which maight be an early indication of multiple-organ failure. Excessive innate immune responses and failure of adaptive immune responses can result in significant morbidity and mortality from SIRS. However, the physiology of inflammation and the pathogenesis of SIRS are highly complex and not fully understood in many aspects, especially regulatory and molecular mechanisms [10].During the examination, we handled the blood samples firstly, then identified the DEGs between SIRS and controls by using bioinformatics analysis.To detect the key genes involved in the process of SIRS, we used the possible hub genes,which highly correlate with the protein–protein interaction (PPI) network to verify the genes.The hub genes of the top two subclusters were identified through the MCODE analysis which shows a high degree of connectivity in the PPI network. The result was listed in Table 4. These hub genes included MX1, OASL, RSAD2, MX2, IRF9, STAT1, OAS1, OAS3, IRF1, IFI44L, IFI44, IRF8, CXCR4, CD86, HLA-DQA2, IL18, CD163, CD68, (IDO1), etc.
The type I or III interferon are the main inducers to enhance the expression of MX family members which can against a diverse range of viruses including pathogens in human and veterinary medicine [21]. Nowadays, more and more DNA and RNA viruses are inhibited by Human MX1.It is essential in defense of mammalian cells against influenza viruses.
Mainly HIV type-1 and other primate lentiviruses are effectively inhibited by Human MX2 which invalid to the MX1-sensitive viruses [22-24]. Therefore, the downregulation of MX1 and MX2 may be a factor in the progression of SIRS.
The 2’–5’-oligoadenylate synthetases (OASs), include OAS1, OAS2, OAS3, and OASL, are a family of interferon (IFN)- and virusinduced proteins. After infected RNA viruses, the proliferation of 2,5-oligoadenylates (2-5A) from ATP was induced by the viral double-stranded (ds) RNA directly binds to, and activates, OAS1-3 [25].Further, the ribonuclease L (RNaseL) , dimerized and activated by 2-5A, can cleave viral and host RNA, so 2-5A plays the most important role in blocking viral replication and inhibiting viral protein synthesis.Recent studies found that OAS1had a negative influence in macrophages about the induction of chemokines and IFN-stimulated genes in response to Toll-like receptor (TLR)3 and TLR4 signaling,whereas in THP-1 cells, OAS3 played a negative role in regulating retinoic-acid-inducible gene I and melanoma differentiation-associated gene 5-dependent antiviral responses. Therefore, the down-regulation of OSA1 and OAS3 would reduce the antiviral capability and accelerate the progression of SIRS.
The IRF family plays an indispensable role in the development and function of natural killer cells.It is necessary to support the rapid growth in the number of mature natural killer (NK) cells when infection and homeostatic proliferation [26]. Therefore, the up-regulation of genes showed a greatly positive role in regulating adaptive immunity.
CXCR4, a 7-transmembrane G protein–coupled receptor, is indispensable in orchestrating both immune responses of innate and adaptive. CXCR4 is critical for the production of T-cell immunological synapse (IS) which may be a driven factor for the initiation of the adaptive immune response [27]. Furthermore, during the process of homing, development, and function of B cells, CXCR4 is indispensable. After infection or injury, the release of large amounts of cytokines, such as IL-2, IL-4, IL-7, IL-10, IL-15, and TGF-β, increases the CXCR4 transcription, thus inducing an excessive immune response and accelerating the development of SIRS.
According to the GO enrichment analysis, most enrichment terms of up-regulated genes were related to the excessive activation and proliferation of immune cells. Following lipopolysaccharide (LPS) stimulation, the multiple downstream protein adaptors and intracellular signaling pathways were activated, producing a large array of cytokines and chemokines [28]. The activation and proliferation of NK cells, lymphocytes, and monocytes are usually promoted by some chemokines, such as CXCR4, IRF8 and so on [24,25]. Recruitment of β-arrestin to the CXCR4–CXCR7 complex activates extracellular regulated kinase 1/2, p38mitogenactivated protein kinase, and stress-activated protein kinase pathways, thus facilitating immune cell migration, survival, and proliferation [29]. IL-6, a dominant inflammatory cytokine, reduces monocyte human leukocyte antigen-DR (HLA-DR) expression and antigen presentation to T cells. It activates C-reactive protein or procalcitonin (PCT) and mobilizes neutrophil progenitors in the bone marrow, leading to peripheral granulocytosis. Its levels are elevated in patients with SIRS [30]. The up-regulated genes enriched in the KEGG pathway were related to antigen processing and presentation, which consistent with hypothesis that excessive activation and proliferation of immune cells may be contained in the regulation of SIRS.
The down-regulated genes were analyzed in the GO and KEGG enrichment. Most of them found to be associated with type I IFN. Usually, type I IFN, through TLR, transfers the LPS signal downstream via interferon-β autocrine induction to induce various inflammatory mediators that exert antibacterial actions. Another study indicated that IFN-α and IFN-β were significant in viral, fungal, and parasitic infections through specific signaling pathways [31]. However, with the progression of SIRS, the over stimulation of the anti-inflammatory and immunosuppressive signals may decrease this response.
There were many accept to improve in this experiment. First, strict screening was not performed. The participants in this clinical trial were not tested for chronic infectious diseases and immune system diseases, such as viral hepatitis, cancer, autoimmune diseases, and gastroenteritis, which might have influenced the results. In addition, the sample size was small, with certain regionality. Hence, it is necessary to make further research through large-sample animal studies.
The purpose of research was to detect hub genes and molecular pathways involved in SIRS via bioinformatics analysis, and thus identify potential diagnostic or therapeutic biomarkers. The excessive activation and proliferation of immune cells and the downregulation of type I IFN genes and TLR signaling pathway were tightly related to SIRS. The verification regarding the relationship of hub genes and function in SIRS needs further studies.
We are grateful to Professor Licheng Zhao from Guangzhou University of Chinese Medicine for his inspiring guidance to this work. We also thank Miss Jingyi Xu from South China Normal University for her critical revision of the article.