Isolation and Purification of Xanthine Oxidase from Discarded Fish Liver Notopterous kapirat: An Easy Source of Enzyme
Xanthine oxidase (XO) has clinical applications as it is an integral part in superoxide dismutase (SOD) diagnostic kit. This investigation deals with isolation, purification and determination of kinetic parameters from fish liver xanthine oxidase. The fractions showing xanthine oxidase activity from fish liver was obtained by heat treatment. Activity staining of a dialyzed sample for discontinuous polyacrylamide gel electrophoresis (PAGE) with non-denaturating and non-reducing condition was performed for confirmation of enzyme activity and showing exact band coincides with a standard enzyme. Purification of enzyme was done in sequential steps of ammonium sulfate precipitation, dialysis, and DEAE-Cellulose column chromatography. The purified XO reveals 2.52 U/mg protein specific activity which represents 27.33 fold purification. The extracted enzyme shows maximum activity at pH 5.0±0.15 and temperature 30±0.38 °C. The kinetic parameters were determined and the values of Km and Vmax for the fish liver fraction are 2.7 mM and 0.035 respectively. The present study shows that fish liver is an easily available and cheap source of xanthine oxidase.
Keywords: Xanthine oxidase; activity staining of XO; Notopterous kapirat; Fish liver
Xanthine oxidase (EC 1.2.3.22) is widespread among species from bacteria to man, even in the tissues of mammals [1-4]. Xanthine oxidase is known to produce reactive oxygen species such as superoxide anion radicals and hydrogen peroxide as a cellular response [5]. One of the foremost intriguing feature of the xanthine oxidoreductase is that it exists in interconvertible form, as xanthine dehydrogenase which utilizes NAD+ as a coenzyme, and is converted to xanthine oxidase which utilizes O2 as an electron acceptor, an interconvertible form [6-8]. XO is a complex enzyme containing flavin adenine dinucleotide (FAD), molybdenum and iron so collectively known as molybdenum iron-sulfur flavin hydroxylases. XO particularly catalyzes the hydroxylation of hypoxanthine (of purines) to xanthine and then to uric acid. The uric acid has been implicated to contribute the antioxidant capacity of the blood. XO catalyzed oxidation involves molecular oxygen as the physiological electron acceptor [9-12]. The enzyme oxidizes a range of substrates including purines, pyrimidines, pteridines, azo purines, cytochrome C, heterocyclic compounds, oxygen, NAD+, and ferricyanide [9,10]. It has been shown to reduce Cyt b5 and p450 in mammalian hepatic microsomes under anaerobic condition [10]. XO is widely used as a detector for nucleotides, purines, superoxide dismutases, adenosine deaminase, phosphates of blood serum and determination of liver diseases [12]. XO is the principal element of the demonstrative unit for estimation of superoxide dismutase (SOD). This kit is very sensitive in the measurement of SOD activity in blood, serum and plasma which is fundamentally utilized as a part of an analysis, checking and treatment of oxidative anxiety. Oxidative anxiety is engaged with the pathogenesis of cardiovascular pulmonary diseases, cerebral dead tissue, mind injury, hypertension, mutagenesis, atherosclerosis, thrombosis, ischemia-reperfusion, diabetes, aspiratory edema, hypoxia, aggravations, cystic fibrosis, Parkinson ailment, Alzheimer sickness, cancer and acquired immunodeficiency syndrome [11,12,14-18]. Estimation of fish meat freshness is important for the food industries for the manufacture of high-quality products. After the death of fish, the decomposition of ATP (adenosine triphosphate) in the fish meat sets in and ADP (adenosine diphospahte), IMP (inosine 5’ phosphate), HxR (inosine), Hx (hypoxanthine), X (xanthine) and U (uric acid). Whereas IMP is one of the major contributing factors to the pleasant flavor of fresh fish, its degradation product hypoxanthine imparts the bitter “off-taste”. In this degradation process, quantification of hypoxanthine can b used as an indication of the fish freshness. Estimation of fish meat freshness is vital for the sustenance enterprises to make the astounding items. After the death of fish, the decay of an adenosine triphosphate (ATP) in the fish meat sets in and adenosine diphosphate (ADP), adenosine 5’ phosphate (AMP), inosine 5’ phosphate (IMP), inosine (HxR), hypoxanthine (Hx), xanthine (X) and uric corrosive (U). Pixie is one of the major contributing components to the wonderful kind of fish; its debasement item hypoxanthine gives the biting “off-taste”. In this debasement procedure, evaluation of hypoxanthine can be utilized as a sign of the fish freshness [13]. The present study was initiated for isolation of crude fractions of XO from fish liver, to find out the relatively easy source of enzyme considering its medical applications especially in the preparation of the superoxide dismutase diagnostic kit. The enzyme is the main component of the diagnostic kit for superoxide dismutase estimation.
The analytical grade chemicals used for the study were purchased from Sigma Aldrich USA, HIMEDIA India, Merck Life Sciences India.
The biological material, fresh fish was purchased from local market Nanded, Maharashtra, India.
Xanthine oxidase crude fractions were isolated from the fish liver by previously reported standard method [1]. In brief, about 10 g of the fresh fish liver was cut into small pieces using a scissoring blade. These pieces were homogenized for 5 minutes in a homogenizer furnished with Teflon pestle with 5 volumes of 0.01 M Tris-HCl buffer (pH 8.0), containing 1 mM EDTA. The homogenate was rapidly heated in a water bath maintained at the 55 °C temperature for 5 min and then cooled quickly in an ice bath. Amid both the heating and cooling steps the homogenate was mixed properly and then centrifuged at 16000 g for 15 min. The supernatant was collected for further studies..
Solid fine powdered ammonium sulfate was added to the extracted fraction in the supernatant to an initial concentration of 30% saturation. The mixture was centrifuged at 16000 g for 15 minutes and resulting precipitate was discarded. The supernatant was again precipitated to the final concentration of 60% saturation. It was centrifuged at 16000 g for 15 minutes. After centrifugation, the pellet was suspended in 10 ml of 0.05 M Potassium Phosphate buffer containing 0.3 mM EDTA, pH 7.5 [1]. The sample was dialyzed in 100 kDa cut off size dialysis membrane against the same buffer for overnight. The overnight dialyzed 0.5 ml sample was loaded on to the DEAE-cellulose column of 1.5 cm width and 12 cm in length previously equilibrated with the 0.2M Phosphate buffer pH 7.4. A column was washed with the same buffer and the enzyme was eluted with an increasing linear gradient concentration ranging from 0.1 to 0.5 M NaCl prepared in the Phosphate buffer. The fractions of 4 ml were collected at the flow rate of 1ml per minute and checked for protein content by a Folin-Lowry method. The enzyme assay for fractions was performed [5]. The fractions showing the enzyme activity were pooled and used for further study.
An activity of XO is evaluated by quantifying the uric acid formation by measuring the absorbance at 290 nm [5]. The reaction mixture containing 0.1 ml extracted enzyme, 1 ml of substrate (xanthine, 50 μM final concentration), and 1.9 ml of sodium phosphate buffer (pH 7.8, 50mM) was incubated for 5 minutes. The time-dependant increase in absorbance was recorded at 290 nm while incubation of the reaction mixture at 25 or 37 °C. One unit of enzyme activity is defined as the rate of formation of one μM of uric acid per minute; the enzymatic activity was calculated by using the formula.
Units/mg protein= (ΔA/min×1000) / (1.22×104 ×mg mL-1 reaction mixture)
Activity staining was performed for xanthine oxidase in the extracted fraction after dialysis by following methods.
Polyacrylamide gel electrophoresis: It was done with the method reported previously [7]. Discontinuous PAGE was performed under nondenaturing and nonreducing conditions, basically as portrayed by Laemmli for denaturing gel. Here, sodium dodecyl sulfate was discarded. Gel dimensions and thicknesses were 100 and 0.75 mm, respectively. Six percent separator gel, without stacking gel, was prepared. After application of the sample, a steady voltage of 100 volts was applied and electrophoretic separation was carried out at room temperature (37 °C). Gels were stained for activity, followed by electrophoresis.
Xanthine oxidase staining in polyacrylamide gels: This method was carried out with slight modifications of the method developed for ascorbate peroxidase [19]. Activity staining on polyacrylamide gels was done at room temperature (37 °C). The mixture of reaction contained 50 mM Tris/HCl, pH 7.6, 0.50 mM xanthine, 0.25 nitroblue tetrazolium and 30 mM TEMED. Staining of the gel was proceeded overnight until the band shows up on the gel. The gel was washed 5-6 times with distilled water for visualization of bands on a gel for both standard enzyme and dialyzed enzyme sample.
Protein determination: Protein content at each step of purification of xanthine oxidase fraction was determined spectrophotometrically according to the Folin-Lowry method by using bovine serum albumin as the standard [20].
The effect of pH on XO activity was studied at various pH ranging from 3.0 to 8.0 with different buffers viz., Citrate- buffer (pH3-5) and Phosphate buffer (pH 6-8). To determine the stability, the enzyme was incubated with different relevant buffers at 30 °C for 10 minutes. The enzyme activities were measured spectrophotometrically at 290nm.
The effect of temperature was determined by incubating the enzyme and substrate samples at different temperatures ranging from 20 °C to 70 °C for 10 mins, and the activity of the enzyme was measured by spectrophotometrically at 290 nm.
The effect of substrate concentration was tested with different concentrations i.e. 4, 8, 12, 16, 20, 24, 28, 32, 36, 40μg/ml by incubating with purified enzyme for 10 minutes at 30 °C. The enzyme activity was measured at 290 nm.
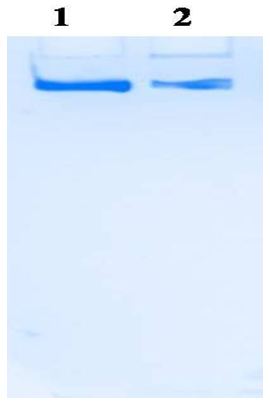
Xanthine oxidase is a commercially important enzyme with a wide area of applications [12]. In the present study Xanthine oxidase was extracted and partially purified from the fish liver by heat treatment, ammonium sulfate precipitation; dialysis and ion exchange chromatography. Activity staining was done with the simple and sensitive method suggested on 6 % separator gel without stacking gel omitting SDS [7]. In Figure 1, lane 1 is the standard enzyme and lane 2 is dialyzed enzyme sample.
It was kept overnight till the appearance of the bands. The electrophoretic analysis of dialyzed enzyme fraction band coincides with, the standard enzyme Xanthine Oxidase purchased from Hi-Media, Mumbai. This activity staining method is satisfactory to confirm the presence of XO comparing with the standard enzyme.
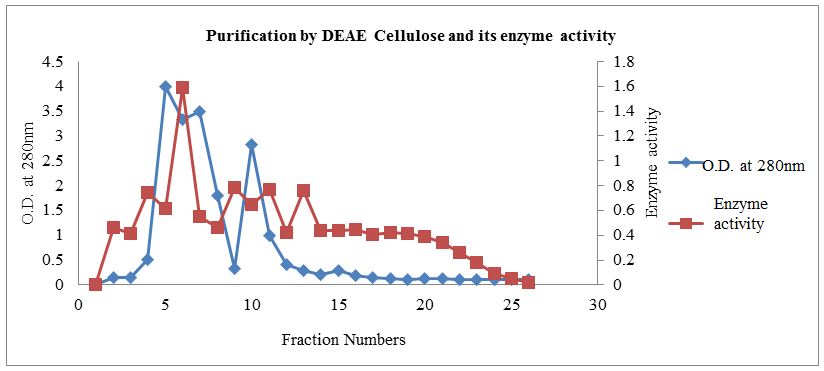
DEAE cellulose column chromatography profile was shown in Figure 2. About 0.1 to 0.5 M NaCl fraction in Potassium Phosphate buffer pH 7.5 was used for elution.Figure 2 revealed the presence of enzyme peak eluted with 0.2 M NaCl fraction. Other 0.1 M and 0.3 M NaCl fractions also showed presence of a protein but without enzyme activity indicating the presence of other protein. Enzyme activity and specific activity on DEAE-cellulose column chromatography were observed to be 0.3666 U/ml/min and 2.5282 respectively.
The Table 1 indicates the good amount of enzyme extracted, the protein content and specific activity up to the dialysis 157 μg/ml and 1.2840 U/mg respectively. Further XO purification is done by DEAE-cellulose column chromatography and 27.33 fold purification obtained with 145 μg/ml protein and 2.5282 U/mg specific activity.
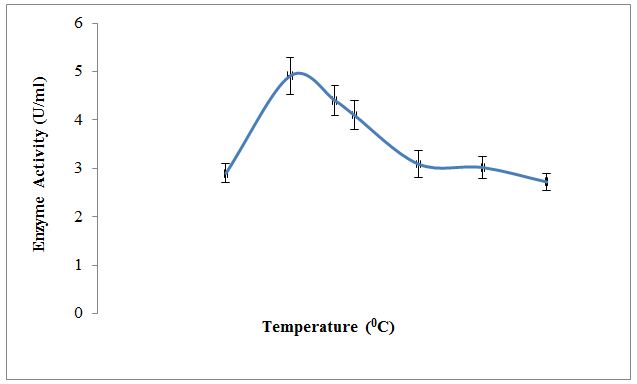
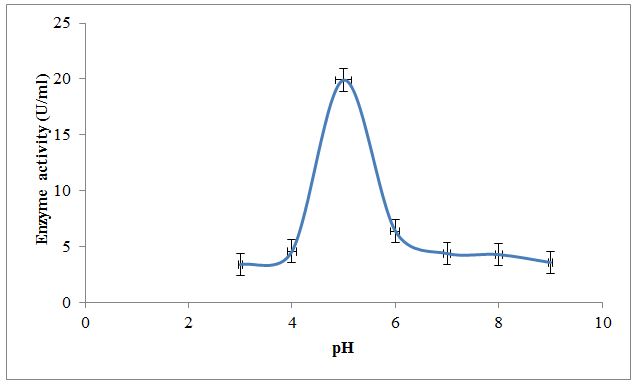
Effect of temperature; pH and substrate concentration on partially purified enzyme along with kinetic parameters like Km and Vmax were also studied. Results in Figure 3 depict the effect of temperature on XO activity. An enzyme is active over the wide temperature range. It was observed that temperature 30±0.38 °C shows the optimum conversion of xanthine to uric acid by an enzyme. Below and above this temperature enzyme activity was lowered. Results in Figure 4 depict the effect of pH on XO activity. An enzyme is active over the wide pH range. It was observed that pH 5.0±0.15 shows the optimum conversion of xanthine to uric acid indicating optimum activity of an enzyme in the acidic pH. Below and above this pH, enzyme activity was lowered. The purified XO shows optimum activity at temperature 30±0.38 °C and pH 5.0±0.15. This may be due to the effect of temperature and pH on active site of enzyme which at temperature and pH favors the binding of substrate. Above and below this temperature and pH binding of substrate to the enzyme active site may be less favorable due to denaturation or conformational changes in the enzyme structure; thereby reducing activity ultimately lowers the optimum conversion of substrate to product [27,28].
Comparing these parameters to other sources of the enzyme like cow and goat milks shows considerable variations. The optimum temperatures for xanthine oxidase from cow and goat milk are 10 °C and 20 °C; however, the value for optimum pH observed for cow milk is 7.5 and goat milk sample ranges from 7.2-7.4 [10]. The temperature and pH optima for desi cows are 25 °C and 7.4 [26]. The effect of pH on buffalo milk XO activity was measured and the most stable pH range for the enzyme activity was found between pH 7.6 – 8.4. The optimum activity of buffalo milk XO enzyme was found at pH 8.0 [9].
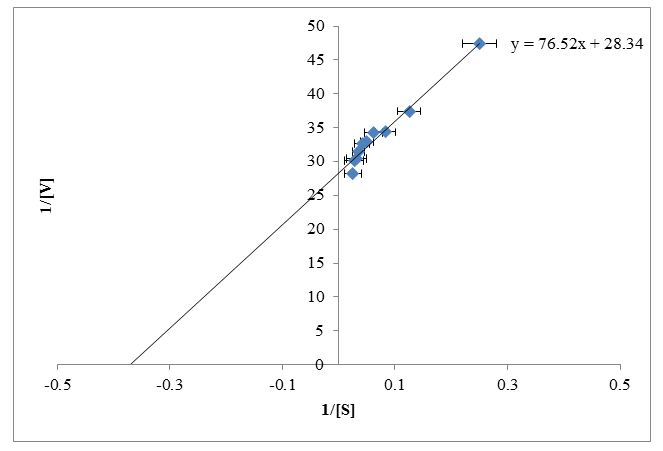
The enzyme kinetic parameters Km and Vmax were determined for the conversion of xanthine to uric acid for fish liver fraction by using the double reciprocal plot. Lineweaver-Burk plot was constructed by using reciprocal of reaction velocity and reciprocal of substrate concentration (Figure 5). Trendline equation y = mx + c was generated from the double reciprocal plot for a straight line in the excel sheet. The values were obtained by using trendline y = 76.52x + 28.34 as shown in Figure 5. Putting the values of slope m = Km/Vmax and C = 1/Vmax, the calculated values for Km and Vmax were 2.7 mM and 0.035 respectively.
XO was also purified from the liver of water buffalo, mouse, rat, rabbit, guinea pig and also from the human. High XO activity was detected in rat and mouse liver whereas XO activity was absent in rabbit and guinea pig. The values for Km and Vmax obtained by Lineweaver-Burk plot by HPLC and spectroscopic method, the Km value for rat are 13.8 and 9.71 while Vmax values for rat are 2.55 and 2.0 whereas the Km value for mouse are 9.67 and 12.0 while Vmax values for mouse are 2.15 and 1.60 respectively and HPLC method was more precise [1].
XO was purified from mouse liver with 302 fold, from rabbit liver with 330 fold, from rat liver with 1200 fold and from human liver 2000 fold [2,22-24]. The XO pH profile from buffalo liver, rabbit liver displayed its optimum activity at pH 7.6, pH 8.1 [12,23]. The value for isoelectric point reported for mouse liver XO as 6.7, and for rat liver XO as 6.13 [22,24]. Reciprocal of the reaction velocity (1/v) and substrate concentration (1/[S]) in Lineweaver-Burk plot when calculated the Km values were 1.1 mM for buffalo liver XO, Km value was 3.4 μM for Mouse liver XO, Km value was 22 μM for rabbit liver XO, Km value was 53 μM for rat liver XO [12,22,23,25]. Low Km value indicates high affinity towards enzyme. Km value for the fish liver is relatively lower as compared to rat, mouse, and rabbit indicating high affinity towards enzyme.
All the above results revealed that XO is showing optimum activity under normal temperature, pH and lower Km value makes it probably a better candidate to serve the purpose. Even dialyzed enzyme sample shows exactly equal and good stained band compared to the standard enzyme purchase from Himedia. As the enzyme is an integral part of SOD diagnostic kit in the medical application; therefore, the purified XO is an easily available and cheap source of an enzyme from the fish liver as it is thrown away after fish processing for food.
This study agrees with earlier reports that xanthine oxidase from different species shows different characteristics. It is very important to study isolation, purification, and characterization of the enzyme as it is an integral part of the diagnostics kit available in the market; hence it is necessary to find out a cheap and the easiest source of the enzyme. The purified enzyme of fish liver very effectively serves this purpose as it is disposed of after fish processing for food preparation. In addition to this, levels of this enzyme in the fish liver also act as a biosensor for water pollution. This property also remarkably influence and promotes the present study.
Authors are thankful to the Director, School of Life Sciences, S R T M University, Nanded for providing necessary facilities. Sagar Jadhao acknowledges UGC for the grant of Maulana Azad National Fellowship for completion of the work.