Methylation Status and Human Age at Three Autosomal Loci: A New Forensic Profiling Tool
Existing criminal DNA profiling methods require prior inclusion of a profile within a criminal database. The ability to obtain descriptive information about an offender from DNA, regardless of database inclusion, would be of great use for investigators. It has been shown that the methylation status of certain human DNA loci correlates with aging. Thus we have used the Illumina MiSeq next generation sequencing platform to investigate numerous genes suggested to have age correlated methylation levels in blood samples from 82 women aged 18 to 91 years. Methylation levels at three CpGs located in ASPA, ITGA2B and PDE4C genes showed an epigenetic signature of aging with a mean absolute deviation of only 6 years from chronological age-an entirely independent confirmation of an earlier study that identified these loci. The implementation of the methylation pattern of these specific autosomal gene sequences from biological evidence left at crime scene may help build up an accurate picture of an offender’s age.
Keywords: Forensic Age Estimation; Methylation Status; Illumina MiSeq Next Generation Sequencing
DNA profiling with numerous highly polymorphic autosomal short tandem repeat (STR) markers has been used in many different aspects of human identification in forensic investigation over the past 20 years [1]. The DNA that is left behind by a criminal at the scene of a crime is useful only when it matches a DNA database or there is reference profile. Scientists are now investigating additional, database-independent, information that can be derived from genotype information in order to generate a description of a suspect [2]. This might predict the gender, physical features of an individual (eye or hair colour) or might suggest the ethnic origin (using ancestry informative markers: AIMs) thus accelerating and targeting the investigative process.
The observation that in all cells the DNA sequence is identical but there are quantifiable epigenetic differences between tissues and across life experiences, suggests another direction for forensic profiling. When cytosine occurs next to guanine in the context of the CG dinucleotide (5-CpG-3) it has the capacity to be methylated on the cytosine to form 5 methyl-cytosine (5mC) [3]. In human DNA 3-6% of all cytosines are thought to be methylated [4, 5]. The forward reaction of methylation is mediated by the DNA methyltransferase enzymes using S-adenosylmethionine as a donor of the methyl group. Demethylation is the backward reaction of methylation which is performed by DNA demethylase enzymes [6]. In addition to 5-mc, 5-hydroxymethyl cytosine (5-hmC) is another cytosine-derived base modification detected in DNA. It is a further derivation from (5mC) catalysed by ten-eleven translocation (TET) enzymes [7].
Studies have shown that different methylation patterns exist across a set of CpG sites in genomic DNA isolated from different tissues (blood, saliva, semen and epithelial tissue) [8], some of which have been termed tissue-specific differentially-methylated regions (tDMRs). This proves experimentally that DNA methylation is a valuable indicator to distinguish body fluids that could contribute to forensic analysis [9, 10]. In other forensic applications, the analysis of DNA methylation patterns may assist with determining the gender of the sample [11], the cause and circumstances of death, the parental origin of alleles, authentication of DNA samples, discrimination of monozygotic twins, and the age of the individual involved [12-15].
Human aging is very complex, multifactorial process and affects all tissues of our body. Various age-indicative markers have been described such as morphological changes of skeletons, teeth and molecular or biochemical changes [16,17]. Increasing age correlates with accumulated genetic changes such as mitochondrial DNA deletions, T-cell receptor deletion and a reduction in telomere length associated with successive cell division, as well as biochemical changes such as the racemisation of aspartic acid and increase in glycation end products. However, all of these approaches suffer from high levels of variation and there are many examples of confounders, such as certain disease states, sex differences, and the origin of the population under study.
The DNA methylation status in a sample has been shown to be affected by the age of the donor. Generally the methylation component will decrease through a life time; so that a centenarian has a lower overall DNA methylation component compared to the DNA of a new-born [18], while the reverse situation is true in gene promoter regions. Claimed that a prediction of biological age through methylation of ELOVL2 gene marker might be used in forensic science with an average of only 7 years error between chronological and predicted age. Blood samples of 303 individual aged 2-75 were used in the study [19].
Here, we describe how DNA obtained from female blood samples was used to interrogate methylation at multiple loci as an epigenetic marker for the prediction of age for forensic purposes.
1. Assess correlations between the chronological age of a person and the methylation state at a number of selected autosomal loci and to validate the results with published studies within the human genome.
2. Determine the optimal experimental techniques (Illumina Sequencing) for extending the sensitivity of the approach so that samples of limited DNA concentration may be tested.
This research project was approved by the University of Strathclyde Ethics Committee and prior to sample donation, participants (All females and from Kurdish Population) signed informed consent statement. The protocol for double swabbing technique (for blood) was followed [20]. The extracted DNA was measurement by qPCR has been previously described [21]. For each reaction conditions a precise DNA concentration was used (120ng) for the bisulphite conversion step, EZ DNA Methylation-DirectTM Kit (Cat # 5020 and 5021, Zymo Research) was applied according to the manufacturer’s protocol [22].
The assembly of genome reference consortium human Build38 (GRCH38 or hg38) was used. The GRCh38 assembly offers an “analysis set” that was created to accommodate next generation sequencing read alignment pipelines. Twenty-three promoters were assessed requiring 50 amplicons which were chosen based on genes previously identified as showing age-dependent methylation changes (Table 1).
Assays were designed targeting CpG sites in the specified ROI using primers created with Rosefinch, Zymo Research’s proprietary sodium bisulphite converted DNA-specific primer design tool. Within this study, parameters were chosen such that PCR amplicons would ideally be bigger than 100 bp but smaller than 300 bp, as described at (http://www.zymoresearch.com/tools/bisulphite-primer-seeker). In addition, these primers were designed such that they would avoid annealing to CpG sites at the region of interest to the maximum extent possible. In the event that CpG sites within primer-binding sites were absolutely necessary for target amplification, additional primers were synthesized with a pyrimidine (C or T) at the CpG cytosine in the forward primer, or a purine (A or G) in the reverse primer to minimise amplification bias of either a methylated or un-methylated allele.
Two pairs of primers were picked to amplify each target region. One pair was used to sequence DNA before bisulphite sodium treatment (Table 2), the other pair was used to amplify chemically converted DNA. Twenty-eight of the 50 amplicons passed the quality control criteria after DNA conversion. The sequences of the primers (after bisulphite treatment) and the estimated amplicon are shown in Supplementary 1.
All primers were resuspended or purchased already in TE solution at 100 μM. Primers were then mixed and diluted to 2 μM. All primers were then tested using Real-Time PCR (Stratagene Mx3005P) with 1 ng of bisulphite-converted control DNA, in duplicate reactions. DNA melt analysis was performed to confirm the presence of a specific PCR product. The following guidelines were used to assess performance: cited at (http://www.zymoresearch.com/tools/qmethyl-calculator/single-sample).
• Had average Cytosine-phosphate (Cp) values <40.
• Duplicate Cps do not have a Cp difference >1 within 5% CT value (CV).
• Reached the plateau phase before the run ended at cycle 45.
• Produced melting curves in the expected range for PCR products.
• Duplicate melts had calculated temperatures (Tms) within 10% CV.
Illumina MiSEQ 300 V2 sequencing was used in this study to enable a high throughput DNA assessment of multiple loci. Library preparation and sequencing were carried by Zymo Research Corporation. Multiplex amplification of all samples using region-of-interest (ROI) specific primer pairs and the Fluidigm Access ArrayTM System was performed according the to the manufacturer’s instructions. The resulting amplicons were pooled and barcoded according to the Fluidigm guidelines. After barcoding, samples were purified (ZR-96 DNA Clean & Concentrator™ - ZR, Cat#D4023) and then prepared for massively parallel sequencing using a MiSeq V2 300bp Reagent Kit (cat. # MS-102-2001) and paired-end sequencing protocol used according to the manufacturer’s guidelines. The consensus sequences of the Fluidigm adaptors were as follows:
CS1= 5’-ACACTGACGACATGGTTCTACA-3’.
CS2 = 5’-TACGGTAGCAGAGACTTGGTCT-3’.
The CS1 and CS2 sequences were included in each of the target or locus-specific primers. The Illumina barcodes were then added in the subsequent barcoding step.
Homology analyses of the DNA sequences were performed by Basic Local Alignment Search Tool (BLAST) which is available at NCBI website (http://www.ncbi.nlm.nih.gov/BLAST/). The NEWCPGREPORT program was used to report CpG island (CGI) located in sequence of each interested region available at EMBOSS Website (http://emboss.bioinformatics.nl/). The PROSCAN-program was used to predict promoter regions in the studied fragment sequences and is available at (http://www-bimas.cit.nih.gov/molbio/proscan/).
To confirm the location of the target region for nDNA the UCSC genome browser (http://genome-euro.ucsc.edu/cgi-bin/hgGateway) was utilized with the newest version of Genome Reference Consortium Human Reference 38 (GRCh38) assembly selected [37].
Sequence reads were identified using standard Illumina base-calling software and then analysed using a Zymo Research proprietary analysis pipeline. Low quality nucleotide stretches and adapter sequences were trimmed off during analysis of quality control.
Sequence reads were aligned back to the reference genome using Bismark (http://www.bioinformatics.babraham.ac.uk/projects/bismark/), an aligner optimised for bisulphite sequence data and methylation analysis [38]. Paired-end alignment was used as default thus requiring both read were aligned within a certain distance; otherwise both reads were discarded. Index files were constructed using the Bismark genome preparation command and the entire reference genome. The non-directional parameter was applied while running Bismark. All other parameters were set to default. Nucleotides in primers were trimmed off from amplicons during methylation calling.
DNA methylation profiles from blood samples from 82 women with ages ranging from 18-91 years were generated using the Illumina MiSeq 300 v2 system. Methylation levels were assessed at 50 target regions within 23 aging-related genes, containing thousands of CpG dinucleotides. Some regions were not amenable to bisulphite primer design due to existing in a region of exceptionally high-CpG density, overlapping with repetitive elements, or both. In total, 28 amplicons of a total 50 passed the quality control (QC) criteria of the assay system (Table 3).
After bisulphite conversion, Illumina sequencing detected methylation levels at 11 of the 28 amplicons, with 100-300bp length (while the other 17 amplicons failing to amplify, failing to sequence, and not showing any methylation differences) incorporating a total of 396 CpG sites analysed (Supplementary Table 2).
Supplementary Table 2 is an Excel file containing the read data and CpG methylation calling for all sites across all samples for the project. The methylation ratio (‘meth ratio’) is calculated by using methylated CpG count/total CpG count. Furthermore, the coordinates for each of the detected CpG sites is listed. If methylation at one CpG site was detected in at least one sample with at least 10 reads, the CpG site was listed in the table. For samples where specific CpG methylation was not detected <10 reads), the meth ratio and total CpG count column was left blank.
Methylation data were installed as tracks on the UCSC Genome Browser for analysis. Evidence for a linear correlation between methylation levels and age was tested by Pearson correlation as detailed in Table 4.
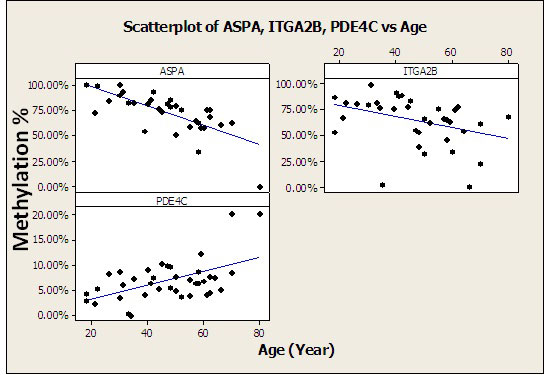
Methylation profiles of eight of the 11 predicted age-related CpG (AR-CpG) sites showed weak association with aging, whereas a subset of three CpGs within ITGA2, ASPA and PDE4C, (coordinates Ch17: 44,390,357, Ch17:3476272 and Ch19:18,233,090 respectively) showed significant correlation with age. Generally, the human genome is hypomethylated during aging [27] with newborn DNA having 494,595 more methylated CpG (mCpGs) dinucleotide than centenarian DNA (16,280,495 vs. 16,775,090 on the Watson and Crick strand, respectively) [18]. By contrast, promoter CpG island methylation levels tend to increase with ageing. These three regions showing greatest correlation with age displayed differing trends: ASPA and ITGA2B were hypomethylated, while the target region in PDE4C was hypermethylated, with aging (Figure 1). These results are consistent with those of other recent studies and suggest that age-related CpG sites (AR-CpGs) are either hypomethylated or hypermethylated [32, 34, 35].
Based on the CpG sites of three genes (ITGA2B, PDE4C and ASPA) a multivariate linear equation was built from a training set of 41 samples by Pearson correlation to predict age of the DNA donors. The resulting equation (Age = 84.93 - 51.92 ASPA+65.2 - 27.4 ITGA2B+34.37 + 189.5 PDE4C) correlated well with chronological age. The mean absolute deviation (MAD) between real age and estimated age was only 5.3 years. Recently Bocklandt and co-workers studied 34 twins aged 21 to 55 years, using DNA from saliva samples [26]. Illumina human methylation27 microarrays were used and, although they were unable to validate the study in different set of samples, a regression model was built using three CpG sites in three genes, NPTX2, EDARADD and TOM1L1 to determine the age of the donor to a mean accuracy of 5.2 years [26].
Koch and Wagner used publically available data (from Human Methylation27 BeadChip analysis) of thirteen tissue types. They found 431 hypermethylated and 25 hypomethylated CpG sites that correlated with age [28]. From these, CpG sites of five genes (TRIM58, KCNQ1DN, NPTX2, BIRC4BP and GRIA2) were able to generate an “Epigenetic-Age” signature with a precision of 11 years.
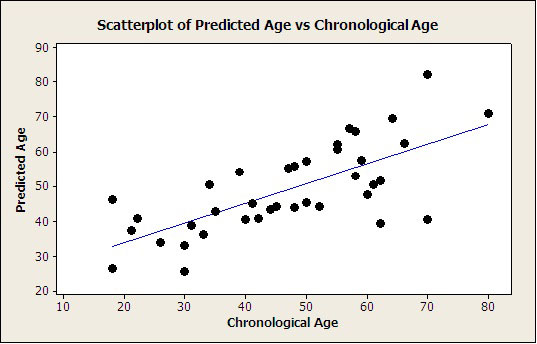
The multivariate linear equation model was validated in a second group of DNA samples (also 41 samples). There was a clear correlation between predicted and real ages (Pearson correlation 0.711 and p-value = 0.0000004); Figure 2, and the MAD was 6.0 years.
It is possible to examine the DNA methylation status of a sample and correlate it with the age of the donor. Our result is similar to the study of Weidner and colleagues who described an age prediction accuracy level of 5 years [29] : differences may be due to the two studies using different assays and DNA obtained from different populations.
Variation in epigenetic age prediction might be influenced by differences in the cellular composition in blood that result from aging [36]. It is still not known if this analysis reflects biological age of the organism or rather of the hematopoietic system [29].
Our findings clearly provide further support that the most promising biomarker predictor of age is DNA methylation modification at specific cytosines – a process that occurs throughout the mammalian lifetime. It is not yet clear how AR-DNAm changes, which seem to occur in a coordinated and reversible manner, are governed and if they have biological consequences.
The precision of the method used here may enable forensic scientists to estimate the age of perpetrators, especially if the assay could be scaled down to work with small trace evidence of DNA. Furthermore, blood evidence is very common at crime scenes and, as DNA is relatively stable, the age prediction approach might enable scientists to investigate even the oldest forensic samples that had been preserved.
The study reported in this chapter has several limitations that have to be taken into account in data interpretation. The methylation measurement was limited to only females, blood samples (a complex mix of white blood cells) and from the same ethnic background (Kurdish). Further studies are needed to isolate nDNA and analyse its methylation changes in specific subtypes of blood cells, other genes, (ELOVL2, for instance) and different tissues (such as saliva, skin and sperm). Further, DNA analysis from other populations and male samples needs to be carried out to assess the generality of our findings. Finally, the impact of epigenetic modifications on determinants of ill health, including smoking, pollution, obesity and chronic life stress should be studied to quantify external influences.
Overall, this experiment has taken a broad look at nDNA methylation pattern in blood samples of different age (18-91 yr). Several interesting gene promotors have been studied. The data presented suggests that the use of methylation modification at three genes (ASPA, ITGA2B and PDE4C) can be used as an indicator of age prediction. The outcomes of this work are applicable not only in forensic biology, but also in clinical research where it might aid the study the molecular basis of the phenomenon of ageing in health and disease.
The authors thank the Ministry of Planning/Kurdistan Regional Government for their financial support in this scholarship as a part of Human Capacity Development Programme (HCDP). We are grateful to all volunteers for providing samples. We acknowledge the Police Forensic Laboratory of Kurdistan/Ministry of Interior for their unconditional support.