Pathogenic variants in KPTN gene identified by Clinical Whole-Genome Sequencing
Status epilepticus is not rare in critically ill intensive care unit patients, but its diagnosis is often delayed or missed. The mortality for convulsive status epilepticus is dependent on the underlying aetiologies and the age of the patients, and thus varies from study to study. In this context, effective molecular diagnosis in a pediatric patient with a genetically heterogeneous phenotype is essential. Homozygous or compound heterozygous variants in KPTN have been recently associated with a syndrome typified by macrocephaly, neurodevelopmental delay, and seizures. To identify the causative genetic defect in a 9 year old male with focal epilepsy, static encephalopathy, autism spectrum disorder, and macrocephaly of unknown etiology who died of status epilepticus. We describe a comprehensive investigation of a patient who was admitted to the intensive care unit. Clinical whole-genome sequencing revealed compound heterozygous variants in the KPTN gene. The first variant is a previously characterized 18-bp in-frame duplication (c.714_731dup) in exon 8, resulting in the protein change p.Met241_Gln246dup. The second variant, c.394+1G>A, affects the splice junction of exon 3. These results are consistent with a diagnosis of autosomal recessive KPTN-related disease. This is the third clinical report for KPTN-deficiency, providing further evidence of a wider range of severity.
Keywords: KPTN; Macrocephaly; Developmental disorders; Intellectual disability; Seizures
List of abbreviations: ACMG: American College of Medical Genetics; DNA: Deoxyribonucleic acid; EEG: Electroencephalogram; OMIM: Online Mendelian Inheritance in Man; MRI: Magnetic resonance imaging; PCR: Polymerase chain reaction; PICU: Pediatric Intensive Care Unit; VLCFA: very long chain fatty acid; WGS: Whole-Genome Sequencing
In recent years, next-generation sequencing (NGS) technologies have revolutionized approaches in clinical genetics. Whole exome or genome sequencing (WES/WGS) allows diagnoses in many patients with complex phenotypes and unusual clinical presentations. As the cost of NGS falls, it has become feasible to use this powerful technology in clinical care, simultaneously unraveling variations in about 19,000 genes. Technological advances have led to the ability to sequence, analyze and interpret entire genome data in a timely manner, clearly changing the diagnostic paradigm and proving to be cost-effective in many cases [1]. Clinical diagnostic sequencing currently focuses on identifying causal mutations in the exome (~1% of the genome), where most disease-causing mutations are known to occur. Whole-genome sequencing (WGS) permits analysis of coding regions as well as regulatory elements that control gene expression, however, non-coding variants remained largely unexplored in clinical diagnostics due to the interpretive challenges [2]. Although it lacks the depth of coverage of an exome, WGS can have more reliable and uniform sequence coverage, particularly in regions of the genome with low sequence complexity or high GC-rich content. Both WGS and WES approaches have benefits and limitations, but the limitation of NGS gene panels and WES should be considered prior clinical testing [1-4]. In this context, the present work demonstrates the application of clinical WGS in the pediatric population where we provided a fast, accurate and cost-effective molecular diagnosis in a pediatric patient with a genetically heterogeneous phenotype.
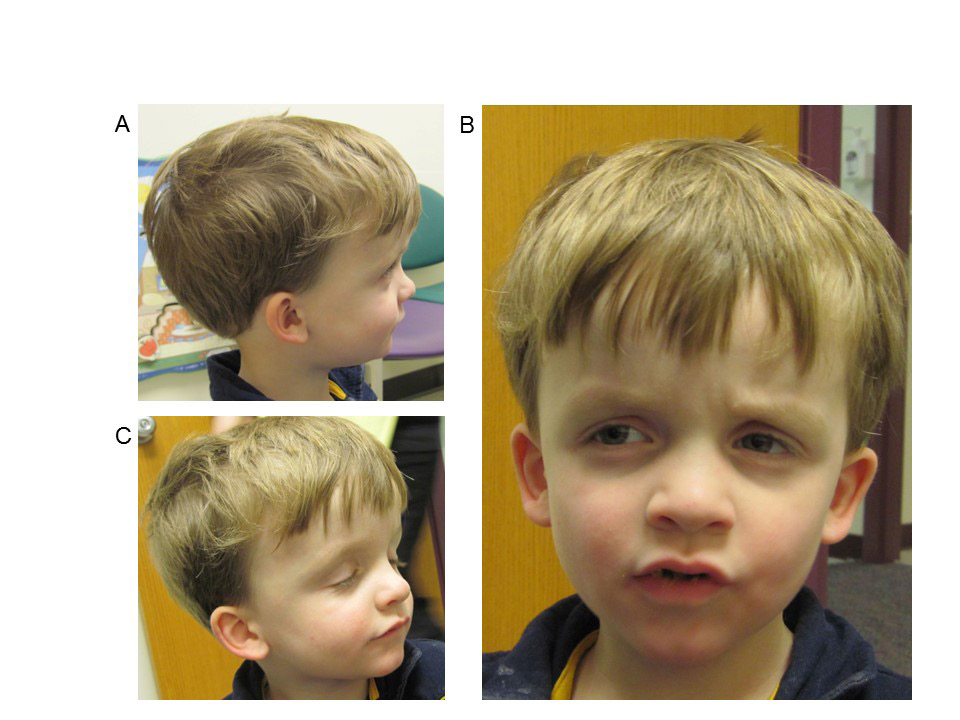
A nine-year-old Caucasian male was admitted to the Children’s Mercy Hospital pediatric intensive care unit (PICU) for status epilepticus and further evaluation of a suspected underlying genetic condition due to his history of macrocephaly, intractable epilepsy, autism, severe developmental delays, hypotonia, and hypoglycemia. Dysmorphic features include frontal bossing, sunken eye sockets, down slanting palpebral fissures, small ears, thin upper lip, and small nose (Figure 1). The prenatal history was unremarkable; delivery was at full term, and neonatal development was normal. He was the third child for his parents together, born to a 26 year old mother and 30 year old father. Family history was noncontributory. At birth, he weighed six pounds nine ounces and was 49.5 cm long. His head circumference was noted to be greater than the 99th percentile at birth. At four years old, he was of average height and weight, but his head circumference measured 57 cm (>98th percentile, +4SD). He had poor tracking and eye contact but a normal ophthalmologic exam. A head CT was completed at two months of age and reported to be normal. At three months of age, he started to have partial seizures but EEG was normal. At five months of age he was noted to have hepatosplenomegaly accompanied by hypoglycemic episodes. Developmentally, he smiled socially at two months, rolled at eight months, sat without assistance at 10-11 months, crawled at 14 months, and walked at two and half years. A brain MRI was normal at four months of age, however a second, performed at 9 years of age, was concerning for a possible arteriovenous malformation, with abnormal hyperintense T2/FLAIR signals in the subcortical white matter of the right anteromedial temporal lobe. An extensive etiologic work-up was normal, including high resolution karyotype, CGH microarray, PTEN sequencing, Fragile X, enzyme analysis for Gaucher, Mucopolysaccharidosis I and II, plasma amino acids, pristanic and phytanic acids, very long chain fatty acid (VLCFA) and urine organic acids. Mild elevations of C14 species on an acylcarnitine profile in 2010 were detected in conjunction with ketotic hypoglycemia and were likely associated with physiologic response to fasting. Previously, the seizures occurred at a frequency of once to twice per hour, consisting of focal tonic clonic seizures involving his left or right side of the body but occasionally progressed to status epilepticus, and a pentobarbital infusion was started with the goal to acquire burst suppression. Unfortunately this was unsuccessful and he passed away at age 9.
Clinical (WGS) was performed on the patient and WES on his healthy mother, following informed consent. Genomic DNA was extracted from peripheral blood mononuclear cells using a Chemagen MSM1 robot (Perkin Elmer, Baesweiler, Germany). WGS was prepared using the Kapa Hyper library prep omitting PCR. Sequencing was completed on an Illumina HiSeq 2500 instrument utilizing paired end 2x125 base pair reads with v4 Chemistry (Illumina, San Diego, CA). The proband’s sample was sequenced to a depth of 115.16 Gb for a mean coverage of ~37x. Bidirectional sequence was assembled, aligned to reference gene sequences based on human genome build GRCh37/UCSC hg19, and analyzed using custom-developed software, RUNES and VIKING [1,5,6]. Variants were filtered to a 1% minor allele frequency, then prioritized by the American College of Medical Genetics (ACMG) categorization, OMIM identity and phenotypic assessment [7]. Alignments were viewed using Integrative Genomic Viewer software version 2.3.8 (IGV; Broad Institute, Cambridge, MA, USA). A candidate gene list was generated by Phenomizer using Human Phenotype Ontology terms: Macrocephaly (HPO:0000256), Muscular hypotonia (HPO:0001252), Autism (HPO:0000717), Seizures (HPO:0001250), Global developmental delay (HPO:0001263), Abnormal facial shape (HPO: 0001999), with a cut off at p value of 0.5 [8]. This gene list contained approximately 700 genes and was imported into VIKING to guide the analysis, however a separate analysis, not limited by this gene list, was performed in parallel to look for relevant pathogenic genotypes in genes not included in the HPO gene list. Pathogenic, likely pathogenic, and variants of unknown significance in HPO genes were reported; likely benign and benign variants are not reported but are made available upon request. Incidental findings in the 56 genes recommended by ACMG were requested by the family and analyzed for pathogenic and likely pathogenic variants only. The patient was compound heterozygous for two pathogenic variants in the KPTN gene. This gene was ranked 351st in the Phenomizer gene list. The first variant identified was an 18-bp in-frame duplication (c.714_731dup ACCGACCACATCTGCAGA; rs587777148) in exon 8, resulting in the protein change p.Met241_Gln246dup. This variant has been previously reported in trans with a second truncating variant in multiple affected individuals from 2 Amish families [9]. Prior in vitro transfection studies indicated the mutant protein is mislocalized and accumulates in neurons of affected individuals, leading to a dominant-negative effect [9]. The p.Met241_Gln246dup variant is not found in population databases such as dbSNP, Exome Variant Database, or ExAC; but was found in 1 of 560 Amish control chromosomes; 136 / 275574 in gnomAD dataset (0.05%) [9]. The second variant, c.394+1G>A, affects the splice junction of exon 3. This variant has not been reported in affected individuals but is predicted to cause aberrant splicing. This variant was observed in approximately ~0.01% individuals of European ancestry in the NHLBI Exome Sequencing Project and Exome Aggregation Consortium (ExAC); 19 / 277090 in gnomAD dataset (0.007%). The c.394+1G>A variant, but not the c.714_731dup variant, was inherited from the mother, indicating these variants are in trans. This genotype was confirmed by Sanger sequencing and is consistent with a diagnosis of autosomal recessive KPTN-related disease. In addition, 2 variants of unknown significance were reported. No incidental findings were reported for this case.
The use of NGS techniques by clinical laboratories has risen tremendously, and have greatly facilitated the elucidation of the etiologic diagnosis in patients suspected of having a genetic disease. The various approaches, including WES, WGS, and targeted panels all have benefits and limitations. Targeted panels offer a specific list of genes relevant to the clinical phenotype in question and often boast higher coverage than WES/WGS. However, whether this improves sensitivity, particularly in Mendelian disorders, is not clear. In addition, a wide range of inter laboratory variability exists for gene lists offered for the same condition. For instance, commercial clinical NGS panels for macrocephaly, include anywhere from 12 to 44 genes. While targeted panels may appear to be a more economical approach to WES/WGS, this is only the case if the gene relevant to the patient being tested is present on the panel, which is often difficult to know a priori, particularly in patients with nonspecific symptoms such as intellectual disability or seizures. In many cases, serial testing of additional genes and panels quickly surpasses the expense of WES/WGS [1]. In addition, targeted panels may fail to incorporate newly discovered disease-associated genes. For example, new genes associated with both syndromic and nonsyndromic macrocephaly have been identified, further expanding the genetic heterogeneity to more than 165 conditions, including 17 metabolic disorders, associated with macrocephaly [4,10-15]. One such gene, KPTN, encoding kaptin, was first reported in January of 2014 in patients with a syndrome typified by macrocephaly, neurodevelopmental delay, and seizures (MRT41; OMIM# 615637) [9,16]. KPTN is not currently offered in any clinical panels for macrocephaly in the USA. Of the clinical laboratories listed in the NextGxDx website, only 2 of 16 offer KPTN as part of an autism/intellectual disability panel, and only 2 of 17 include it as part of an epilepsy panel. The comparison of such gene lists is difficult for clinicians and the curation is onerous for the clinical laboratory to manage. Clinical WES/WGS removes the guesswork as far as which gene to include, as all genes relevant to the patient’s phenotype are queried in the analysis process. In the current case, by using clinical WGS in a nine-year-old Caucasian male admitted in pediatric intensive care unit for status epilepticus, a diagnosis of KPTN-related disease was made.
A comparison of the individual clinical signs in patients reported with KPTN-related disease are shown in Table 1. The most unifying features are macrocephaly (92%), developmental delay (100%) and intellectual disability (100%). Recurrent dysmorphic features included frontal bossing, abnormal head shape and prominent chin. With the exception of the p.Met241_Gln246dup, which is predicted to be nonfunctional by producing a misfolded altered protein, all patients have truncating variants; no patients were homozygous for the p.Met241_Gln246dup. RT-PCR or western blot experiments were not performed to assess if those variants result in nonsense-mediated mRNA decay or whether a truncated protein (lacking the C-terminal amino acids) is produced. The second variant uncovered in our patient, c.394+1G>A, is located upstream of the two previously reported truncating variants. This variant is more likely to result in nonsense-mediated mRNA decay, which may explain the more severe outcome. Additional external factors such as modifier genes could also influence the phenotype. Of the 11 previous patients described, seven have survived into their second and third decades, with two having expired at ages 29 and 30 to a head injury and pneumonia (Table 1).
This is the third clinical report for KPTN-deficiency, and the first to die in childhood with status epilepticus, providing further evidence of a wider range of severity.
Several studies have shed new light on the molecular and cellular processes that orchestrate the human neuronal circuitry and is defective in neurological disorders [9,11,12,17,18]. Evidence implicating defects in proteins involved in actin dynamics, microtubule homeostasis, and vesicle trafficking in developmental disorders via effects on neuronal development and migration and brain architecture has accumulated over the past decade. The role of microtubule homeostasis (LIS1 and DCX) or tubulin (TUBA1A, TUBA8, TUBB2A, TUBB4A, TUBB2B, TUBB3, and TUBB and TBCE) genes has been established in several brain disorders. More recently, the actin-encoding genes ACTB and ACTG1 have been shown to be involved in brain malformations causing Baraitser–Winter syndrome (OMIM # 243310) [17]. Additional intellectual disability genes involved in actin dynamics and vesicle trafficking include STXBP1 and SYP [19,20]. Patients with KPTN-deficiency consistently exhibit macrocephaly, intellectual disability and developmental delay. Furthermore, KPTN is a novel actin binding protein, enriched in neuronal growth cones and cortical sites of neurons at early developmental stages, likely playing a central role in modulating neuron morphology and growth. Although genes affecting many of these pathways are associated with hearing loss, to date, no patients with KTPN-related disease, including our patient, have been reported with deafness. As with several other published studies, whole-genome sequencing was found to be a rapid and cost-efficient approach for molecular diagnostic of a genetically heterogeneous condition [1-3,6,17]. Thus, this report confirms the occurrence of KPTN-related syndrome outside of the Amish population and demonstrates the variability in the phenotypic spectrum and severity, even with the same pathogenic variant. Pathological mechanisms of abnormal neuronal actin cytoskeleton and discrepancy between the underlying phenotypes caused by KPTN deficiency remain to be elucidated.
We are very grateful to this family for allowing this case to be published. We thank our colleagues in the Center for Pediatric Genomic Medicine and Children’s Mercy Hospital in Kansas City. This work was not support by grant
The author(s) have made the following declarations about their contributions: Conceived and designed the experiments: CJS, EF, IT. Performed the experiments: CJS, EF, IT, LZ, SS. Contributed reagents/materials/analysis tools: NM, EF. Wrote the paper: IT, CJS. Reviewed the manuscript: EF,SS, LZ, BH, AA, NM. Contributed to the recruitment and clinical investigations of the patient for the study: AA, BH, AA.
The accession numbers for the reference sequences are listed as follows:,
KPTN; NM_007059.2;
1000 Genomes Project, http://www.1000genomes.org/
NHBLI Exome Sequencing Project (ESP) Exome Variant Server,
http://evs.gs.washington.edu/EVS/
ExAC Browser (Beta) | Exome Aggregation Consortium; http://exac.broadinstitute.org/
IGV; https://www.broadinstitute.org/igv/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
SNPdb, http://www.ncbi.nlm.nih.gov/projects/SNP
PolyPhen-2, http://www.genetics.bwh.harvard.edu/pph2/
ClinSeq, http://genome.gov/20519355
RefSeq, http://www-ncbi-nlm-nih-gov.proxy2.library.mcgill.ca/RefSeq
SIFT, sift.jcvi.org/