Postpartum Dilated Cardiomyopathy, the Role of Genes
We presented a 28 year old woman presenting a postpartum cardiomyopathy who required heart transplantation. Interestingly; her mother presented 2 years after a dilated cardiomyopathy so they were both screened for Titin (TTN) gene mutations.A heterozygosis of variant in the TTN gene was found.
Keywords: Dilated Cardiomyopathy; Titin Gene; Heart Failure
Peripartum cardiomyopathy has traditionally been considered a non-genetic or familiar type of dilated cardiomyopathy associated to pregnancy [1]. Nowadays this entity is considered as an independent disease itself which diagnoses is made upon a temporal relation with pregnancy and the exclusion of other cardiomyopathies. However, latest investigations suggest the role on inheritance factors in the development of the disease [2].
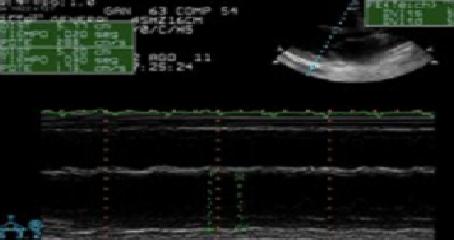
28 year old woman with no previous medical history who recently presented a normal delivery at week 38 of gestation. She was admitted to the emergency department for progressive dyspnea starting two weeks prior to delivery. Upon arrival she was tachycardic and oxygen saturation on room air was 91%. Electrocardiogram showed a sinus tachycardiawith diffuse repolarization changes (Figure 1). Cardiomegaly and signs of vascular redistribution were found on the chest X-rays. A complete laboratory panel showed increase Troponin T (0,03 ng/dL), proBNP (6000 pg/mL) and D Dimers (>5000 mg/dL). Considering a recent delivery as well as the laboratory tests, a thorax CT scan to rule out pulmonary embolism was indicated but it only demonstrated the presence of pleural effusion and vascular congestion. The echocardiogram revealed dilation and severe dysfunction of left ventricle as well as important mitral and triscupid insufficiency (Figure 2). In the setting of this clinical picture, the diagnosis of peripatum cardiomyopathy was evoked.The patient clinically progressed to severe acute heart failure and was transferred to the intensive care unit. She needed orotracheal intubation, noradrenaline and levosimendan. Diuretics, bromocriptin and angiotensin enzyme inhibitors were added. Cardiac magnetic resonance did not show edema or late enhancement so myocarditis was ruled out. Heart catheterization revealed pulmonary capillary pressure 29 mmHg and mean pulmonary artery pressure 35 mmHg accompanied by increased transpulmonary gradient (>12 mmHg ) and cardiac output estimated of 1.6 ml/min. Extracorporeal membrane oxygenation system (veno–arterial) was added to the whole therapeutic arsenal and 24 hours later a counterpulsation balloon was placed. In the absence of clinical improvement after three weeks of treatment, the patient was transferred to a specialized medical center for heart transplantation which was successfully performed 20 days after the initiation of extracorporeal membrane oxygenation therapy.
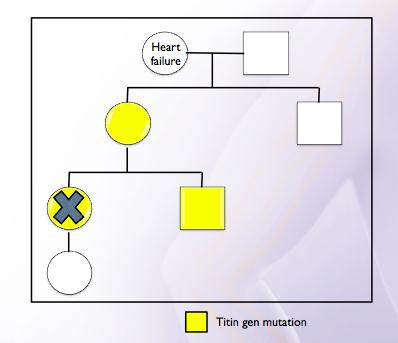
Two years later, the patient’s mother presented with acute heart failure at age 48. Dilatation and severe left ventricle dysfunction were observed in the echocardiogram. No coronary lesions detected on the heart catheterization. Considering the presence of dilated cardiomyopathy in two members of the same family, genetic studies by means of next-generation sequencing (Illumina Genome Analyzer II) were conducted.
In this study, a heterozygosis of variant c.40460_40461insT; P.Leu13487Phefs* 6 in the TTN gene (2q31, which encodes the sarcomeric titin protein) was detected in both patients. The patient’s brother was also screened testing genetically positive but phenotypically negative, it means, the absence of structural cardiomyopathy (Figure 3).
Although the clinical course of peripartum cardiomyopathy is generally benign, with recovery of function and ventricular dimensions [3]. There are a proportion of patients (4-5%) who progresses to refractory heart failure requiring vasoactive support and heart transplantation as in our case. The European Society of Cardiology defines postpartum cardiomyopathy as a non-familiar or genetic dilated myocardiopathy associated to pregnancy [1]. However, recent investigations suggest that hereditary factors might play an important role in the pathogenesis of this disease [2]. They have demonstrated that postpartum cardiomyopathy shares a similar genetic predisposition with idiopatic dilated cardiomyopathy placing mutations of titin gene as the principal disorders in both entities [4].
The mutations of the titin gene located in chromosome 2 accounts for nearly 25% of familiar dilated cardiomyopathy [5]. This gene encodes the protein titin found in the sarcomeres of cardiomyocytes. The main function of this protein is to provide flexibility and stability to these cellular structures, as well as in the chemical signaling process. More than 40 mutations of the titine gene leading to familiar dilated cardiomyopathy have been described. These mutations alter the function of the sarcomeresubsequently leading to dilation and ventricular dysfuntion. Genetic forms of this disease are considered to have less probability to a complete recovery demonstrated by a lower left ejection fraction at one year follow up [5].
Pospartum cardiomyopathy may have a genetic component and may represent the initial manifestation of dilated familar cardiomyopathy so first degree relatives should be screened.
1. Postpartum peripartum cardiomyopathy may have a genetic cause and some patients considered to have a postpartum cardiomyopathy are just a dilated cardiomyopathypresenting after delivery.
2. Postpartum peripartum cardiomyopathy is generally benign but may evolve into refractory heart failure.